
LA DISSECTION ULTIME DES GENES ET LEUR RECOMBINAISON IN VITRO.
1 CARTOGRAPHIE A HAUTE RESOLUTION
1.1 OBTENTION DE MOLECULES D'ADN HOMOGENES
Comme on l'a vu au chapitre "Structure et propriétés des molécules d'ADN", ces polymères représentent des fils extrèmement long pour un diamètre très faible. Une conséquence est qu'au cours de la purification de l'ADN, ces fils subissent des cassures aléatoires de sorte, qu'en termes de séquences, on obtient un mélange héterogène.
Ce sont des nucléases, c'est à dire les enzymes spécialisées dans la dégradation des acides nucléiques qui ont permis de régler le problème du découpage non aléatoire d'un génome in vitro.
Il existe deux catégories de nucléases selon leur mode
d'action
: les exonucléases qui
dégradent
par excision du nucléotide situé en
extrémité
de chaîne (souvent en 3') et les
endonucléases qui coupent des
liaisons
phosphodiester dans la chaîne.
Parmi les endonucléases, certaines sont non spécifiques
et
hydrolysent des liaisons phosphodiester au hasard, d'autres
présentent
une remarquable spécificité de
site.
Un ensemble est constitué d'enzymes spécifiques d'ADN
bicaténaire, capables de reconnaître une séquence
précise (de 4 à 8 nucléotides) et de couper la
molécule avec une précision qui atteint le
nucléotide.
Exemple de sites de reconnaissance et de coupure pour deux endonucléases de restriction dans une même molécule d'ADN.
Remarque : Ces enzymes sont appelées des
endonucléases de restriction car, in
vivo, elles permettent aux bactéries de reconnaître
certains
ADN de bactériophages et de les dégrader, restreignant
ainsi leur capacité de croissance, toutes les espèces
bactériennes disposent ainsi d'une ou plusieurs de ces armes.
Elles
se protègent elles mêmes de l'action de ces enzymes en
modifiant
leur propre ADN aux sites de reconnaissance (également
appelés
sites de restriction). La modification
consiste
souvent en une méthylation de certaines bases contenues dans le
site
de restriction, un ADN exogène, non méthylé, sera
reconnu
et dégradé.
|
Nucléase |
Source (rappelée dans |
Site reconnu |
Nucléase |
Source (rappelée dans |
Site reconnu |
|
Hæ III |
Hæmophilus ægyptus |
Im |
Hpa II |
Hæmophilus parainfleuenzæ |
Im
|
|
Mbo I |
Moraxella bovis |
I
|
Taq I |
Thermus aquaticus |
I
m |
|
Bam HI |
Bacillus amyloliquefaciens |
I
m |
Bgl II |
Bacillus globiggi |
I
|
|
Eco RI |
Escherichia coli |
I m
|
Hin dIII |
Hæmophilus influenzæ |
mI
|
|
Eco RII |
|
I m
|
Pst I |
Providentia stuartii |
I |
(I représente le site de coupure pour chaque brin, m un site de méthylation lorsqu'il est connu).
 fragments
différents, par contre, s'il existe un seul site reconnu par une
enzyme
de restriction donnée, sous l'action de cette enzyme, le
plasmide
va être linéarisé en produisant des
molécules
identiques au nucléotide près, s'il existe deux sites, on
obtiendra
deux fragments de tailles parfaitement définies.
fragments
différents, par contre, s'il existe un seul site reconnu par une
enzyme
de restriction donnée, sous l'action de cette enzyme, le
plasmide
va être linéarisé en produisant des
molécules
identiques au nucléotide près, s'il existe deux sites, on
obtiendra
deux fragments de tailles parfaitement définies.
résultat de 4 coupures aléatoires dans une molécule d'ADN circulaire
résultat d'une coupure ciblée dans une même molécule
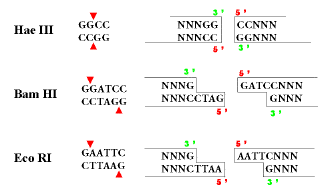
L'examen du tableau précédent montre que tous les sites sont des palindromes (présentent un centre de symétrie), certaines de ces nucléases coupent les deux brins au même niveau (selon l'axe de symétrie) et génèrent des extrémités franches, d'autres d'une façon décalée et génèrent des extrémités débordantes soit en 5' soit (plus rarement) en 3'.
Les extrémités débordantes sont
également
appelées extrémités cohésives car, en
raison
de la structure palindromique
 des
séquences de reconnaissance, elles ont tendance à
s'hybrider
en solution. Cette situation favorise l'action d'une
ligase (une
ligase
est une enzyme capable de créer une liaison phosphodiester entre
deux
chaînes polynucléotidiques), elle sera souvent mise
à
profit dans les recombinaisons in vitro
et la construction de molécules chimères.
des
séquences de reconnaissance, elles ont tendance à
s'hybrider
en solution. Cette situation favorise l'action d'une
ligase (une
ligase
est une enzyme capable de créer une liaison phosphodiester entre
deux
chaînes polynucléotidiques), elle sera souvent mise
à
profit dans les recombinaisons in vitro
et la construction de molécules chimères.
*Remarque : Dans la plupart des cas, les fragments produits sont phosphorylés en 5' ce qui n'est pas toujours la règle lorsque l'on hydrolyse des polynucléotides.
1.2 CARTOGRAPHIE DES GENES A HAUTE RESOLUTION
Comme on l'a vu dans le premier chapitre, la position de certains
gènes
sur le chromosome peut être déterminée en mesurant
la
fréquence de recombinaison entre le gène
étudié
et un marqueur déjà repéré (autre locus).
Cette cartographie est dépendante
d'une
densité suffisante de marqueurs. De plus elle ne nous apprend
rien
sur l'organisation "interne" du cistron : chez un eucaryote
supérieur,
un gène "moyen" (1000 à 5000 pb) représente 0,01
à
0,001 cM, pour détecter un seul événement de
recombinaison
à l'intérieur d'un gène, il faudrait pouvoir
observer
10 000 à 100 000 individus issus d'un même croisement.
Relier un gène à d'autres, en groupes de liaisons n'est donc que le début de la carte génétique. La cartographie ultime consistera à obtenir la séquence complète des nucléotides du gène et de son voisinage ce qui peut représenter un travail considérable. Entre les deux (position du gène sur le chromosome et séquence) il existe des intermédiaires qui font appel à d'autres techniques que l'évaluation de la recombinaison.
1.2.1 CARTES DE RESTRICTION
Pour disséquer le gène on va utiliser les propriétés des nucléases de restriction qui reconnaissent des points précis du génome au nucléotide près. Chaque enzyme de restriction ayant une cible particulière (voir la première figure), l'identification des sites de coupure permet d'établir une carte extrêmement détaillée appelée carte de restriction.
Les points de coupure sont établis d'après la longueur des fragments obtenus après digestion d'un segment d'ADN par une enzyme de restriction donnée. La taille des fragments est elle même déterminée par électrophorèse. Un jeu de fragments de taille connue sert à calibrer le gel et l'on peut estimer d'une façon relativement précise celle des segments générés par une digestion enzymatique.
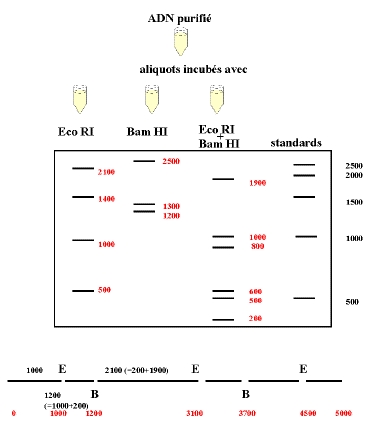 La
figure ci-contre
résume une expérience de cartographie. Dans cet exemple
on
étudie l'ADN préparé à partir d'un
bactériophage fictif dont le génome serait une
molécule
linéaire de 5000 paires de bases.Une digestion avec l'enzyme Eco
R1
permet d'obtenir 4 fragments : 2100, 1400, 1000 et 500 pb , il existe
donc
3 sites de coupure (sites de restriction) pour cette enzyme. Une
expérience
menée en parallèle montre que la molécule de
départ
(5000 pb) possède deux sites de restriction pour Bam H1 (B) le
coupant
en 3 morceaux : 2500, 1300 et 1200 pb.
La
figure ci-contre
résume une expérience de cartographie. Dans cet exemple
on
étudie l'ADN préparé à partir d'un
bactériophage fictif dont le génome serait une
molécule
linéaire de 5000 paires de bases.Une digestion avec l'enzyme Eco
R1
permet d'obtenir 4 fragments : 2100, 1400, 1000 et 500 pb , il existe
donc
3 sites de coupure (sites de restriction) pour cette enzyme. Une
expérience
menée en parallèle montre que la molécule de
départ
(5000 pb) possède deux sites de restriction pour Bam H1 (B) le
coupant
en 3 morceaux : 2500, 1300 et 1200 pb.
Il existe plusieurs moyens de construire une carte à partir de
ces
données. Le premier résulte d'une double digestion : on
va
regarder où sont localisés les sites d'une enzyme dans
les
fragments obtenus par l'autre. Il serait possible d'extraire chaque
bande
du gel d'électrophorèse et de soumettre l'ADN à
une
seconde digestion, pratiquement ce serait très long et
fastidieux.
Il est plus simple d'ajouter les deux enzymes en même temps dans
le
milieu d'incubation. Après digestion complète (tous les
sites
reconnus par l'une ou l'autre enzyme sont coupés),
l'électrophorèse permet de séparer des fragments
de
1900, 1000, 800, 600, 500 et 200 pb. Le raisonnement repose sur
l'additivité parfaite des tailles : si le fragment E 2100
possède
un site B à 200 pb de l'extrémité, les fragments
générés par la double digestion seront de 200 et
1900
pb.
On remarque que les fragments 1000 et 500 se trouvent à la fois
dans
la digestion E et dans la double digestion il s'agit donc des
extrémités. La suite repose sur le chevauchement des
fragments
produits par l'une ou l'autre enzyme, si l'on choisit 1000 pb comme
premier
site de coupure pour E, on doit trouver dans la double digestion un
fragment
n tel que 1000 + n = taille d'un fragment B qui correspond au 1er site
reconnu
par B 200 est candidat et l'on place un site B à 1200 pb de
l'extrémité ... et ainsi de suite.
Lorsque l'on étudie des régions d'ADN de plus grande taille, les profils d'électrophorèse deviennent plus complexes, des fragments de même taille se rencontrent (on ne pourra pas les distinguer car ils migrent au même endroit) on doit donc adopter d'autres stratégies.
Une approche possible consiste à réaliser des
digestions partielles : on régle
les
conditions de concentration d'enzyme, de température, de temps
d'incubation... de telle sorte que statistiquement un site sur deux ou
trois
soit coupé (au hasard)
 On
obtiendra
par exemple, pour une digestion partielle E, des fragments de 3500,
3100
et 1400 pb, en comparant avec les résultats de la digestion
complète, on peut conclure que les fragments 1000 et 2100 sont
contigus,
de même, 2100 et 1400 sont deux fragments adjacents du 3500
etc...
On
obtiendra
par exemple, pour une digestion partielle E, des fragments de 3500,
3100
et 1400 pb, en comparant avec les résultats de la digestion
complète, on peut conclure que les fragments 1000 et 2100 sont
contigus,
de même, 2100 et 1400 sont deux fragments adjacents du 3500
etc...
Le marquage en 5' par du phosphore 32 grâce à la
polynucléotide kinase (enzyme
capable
de phosphoryle des extrémités 5' préalablement
déphosphorylées) peut également rendre des
services
en permettant d'identifier d'emblée le segment terminal. Ici
encore
on utilise deux enzymes, mais successivement, pour obtenir des
fragments
dont une seule extrémité est marquée (après
électrophorèse, une autoradiographie est
nécessaire
pour identifier le fragment d'extrémité).
En combinant le marquage 5' et les digestions partielles, on peut lire
directement la carte sur l'autoradiogramme puisque tous les segments
internes
n'apparaissent pas.
1.2.2 UTILISATION DES SITES DE RESTRICTION COMME MARQUEURS
L'analyse mendélienne repose sur l'existence de
différentes
formes alléliques d'un même
"gène" et passe par l'expression de ceux-ci : il s'agit aussi
bien
de couleurs de fleurs que de la présence d'alloenzymes. La
biologie
moléculaire a permis de montrer que, quel que soit le niveau de
l'observation phénotypique, ce repérage allélique
est
bien en dessous du polymorphisme réel
de l'ADN.
On comprend aisément que les mutations faisant apparaître
des
formes alléliques nouvelles peuvent modifier la carte de
restriction
: une insertion ou une
délétion affectant la
fonction
d'un gène vont provoquer une modification de la taille de
certains
fragments de restriction. Mais la technique va beaucoup plus loin :
elle
permet de discerner un polymorphisme dans les molécules d'ADN
même
s'il n'a aucune incidence phénotypique : dans des codons
synonymes,
dans des introns, dans des séquences
répétées,
dans des séquences non exprimées ...
Bien que sur le plan évolutif ces variations n'aient probablement pas de grandes significations, elles peuvent être exploitées à plusieurs fins :
Le sigle RFLP (restriction fragment length polymorphism) évoque la méthode qui va permettre une analyse allélique directe, au niveau de l'ADN.
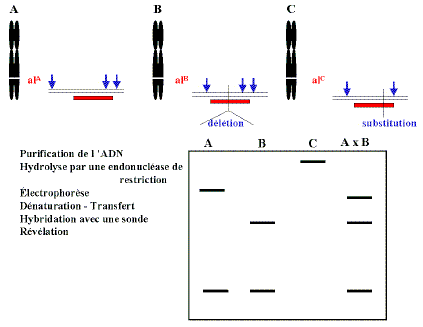
Entre deux génomes, des différences de tailles de fragments d'ADN homologues obtenus par digestion à l'aide d'enzymes de restriction seront observées si des substitutions, des insertions ou délétions ont eu lieu au niveau des sites de reconnaissance de l'enzyme ou si des réarrangements de quelque importance se sont produits entre deux de ces sites de restriction. Dans l'exemple ci-contre, on met en évidence une délétion qui racourcit un fragment (B) et une substitution qui supprime un site (C).
Etant donné que l'organisation en fragments caractéristiques se transmet de façon mendélienne, on peut les utiliser pour repérer des loci de la même façon qu'un caractère morphologique avec un certain nombre d'avantages.
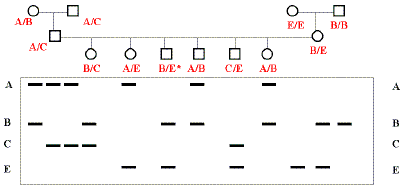
Exemple de transmission mendélienne des marqueurs "RFLP". De l'ADN de chaque individu figurant dans l'arbre généalogique a été digéré par une nucléase de restriction. Les fragments séparés par elecrophorèse sont hybridés avec une sonde. Quatre allèles (A, B, C, E) sont révélés au locus repéré par la sonde.
Pratiquement : On prépare l'ADN
total
de différentes espèces, de variétés voire
même
d'individus. Cet ADN est ensuite incubé en présence de
nucléases de restriction et les produits de digestion
séparés
par électrophorèse. La visualisation de bandes distinctes
après électrophorèse ne peut se faire
qu'après
transfert et hybridation avec une sonde, l'obtention des sondes sera
vue
au chapitre suivant. En effet, la taille d'un génome
eucaryotique
est telle que le nombre de fragments générés et
leur
taille donnent une répartition pseudoaléatoire tout le
long
du gel.
La figure ci-dessous est un autre exemple d'application à la
cartographie
: une sonde donnée a été hybridée aux
fragments
de restriction d'ADN extrait de deux lignées pures de maïs
:
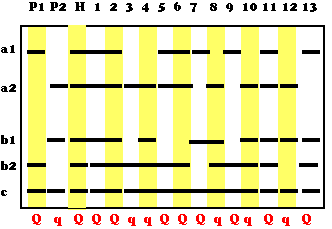
P1 et P2, d'un individu F1 et de 13 individus de la F2.
On constate :
 La
distinction
des allèles et des loci se fait en examinant un certain nombre
d'individus
F2. On voit par exemple que chez le 3, les bandes a1 et b1
sont absentes, elles ne peuvent donc pas représenter les deux
formes
allèliques d'un même locus (confirmé par a2
et
b2 absents en 7). Donc a1 et a2 sont bien deux
allèles
d'un locus et b1 et b2 sont ceux d'un autre locus.
La
distinction
des allèles et des loci se fait en examinant un certain nombre
d'individus
F2. On voit par exemple que chez le 3, les bandes a1 et b1
sont absentes, elles ne peuvent donc pas représenter les deux
formes
allèliques d'un même locus (confirmé par a2
et
b2 absents en 7). Donc a1 et a2 sont bien deux
allèles
d'un locus et b1 et b2 sont ceux d'un autre locus.
La technique mendélienne rappelée au premier chapitre
permet
d'associer ces marqueurs à des éléments
cartographiques
connus (caractère qualitatif, isoenzyme...) ce qui permet
d'établir
des cartes précises de plus en plus denses.
Ceci est précieux dans un programme de sélection : si un
caractère important (appelé Q
dans
la figure) est associé au marqueur A1 par exemple, on
repère
très facilement en F2 les allèles A1 et de prédire
que
le caractère Q sera présent.
| Conclusion : L'analyse directe du polymorphisme de l'ADN se répand très rapidement pour des raisons évidentes :
|
* Remarque : repérage des
caractères
quantitatifs
Les cartes génétiques classiques ne recensent que des
caractères qualitatifs c'est
à
dire des allèles dont la forme mutée se distingue
clairement
sur le plan phénotypique.
Des caractères tels que la taille, le poids, la
résistance
à des agents pathogènes... sont gouvernés par un
nombre
important de gènes, chacun d'eux participe d'une façon
minime
à l'élaboration du phénotype. La mutation de l'un
d'entre
eux a le plus souvent un effet imperceptible qui exclut les
observations
classiques de recombinaison. De plus, leur expression est
particulièrement
sensible aux fluctuations de l'environnement. L'association
étroite
de marqueurs "RFLP" à des loci gouvernant de tels
caractères
permet de les traiter par l'analyse mendélienne.
1.2.3 SEQUENCAGE DES GENES
L'étape ultime de la cartographie est la séquence, non pas du génome mais modestement de petits morceaux intéressants délimités par la cartographie de restriction.
Le principe du séquençage est simple, la réalisation plus ou moins délicate selon les méthodes utilisées.
Il s'agit, en partant du segment à étudier, de
générer
des fragments qui tous ont la même origine et qui tous se
terminent
par un nucléotide de même nature : il faut construire 4
séries
de fragments : tous commencent au même point, une série se
termine
par A, une autre par T, une autre par G et une dernière par C.
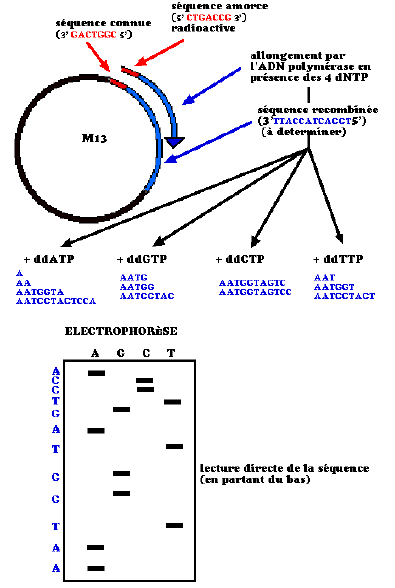
Exemple pratique: On suppose le segment à étudier
cloné
dans un vecteur simple brin (bactériophage M13, cf plus loin).
Connaissant
bien le vecteur, on dispose de petites séquences
complémentaires
de régions proches de l'insertion.
Après hybridation, ces séquences peuvent servir d'amorce
à
une ADN polymérase. L'astuce consiste à réaliser
une
synthèse in vitro en ajoutant aux nucléotides
(dNTP)
nécessaires à la réaction une petite
quantité
de didéoxynucléotides. Si l'un d'entre eux est
incorporé,
la chaîne ne présente plus d'extrémité 3' OH
et
la polymérase s'arrête. On prépare 4
réactions
avec respectivement une petite quantité de ddATP, ddTTP, ddGTP
et
ddCTP. Statistiquement, la réaction s'arrêtera
respectivement
en face de n'importe quel T, A, C et G selon le tube. Les produits de
la
réaction, dans chaque tube, seront des fragments qui tous
commencent
à l'extrémité 3' de l'amorce et qui tous se
terminent
à chaque A possible, T, G ou C. Après séparation
de
ces fragments par électrophorèse en gel de polyacrylamide
dénaturant, les quatre pistes révèlent directement
la
séquence.
D'autres méthodes, utilisant des réactions chimiques spécifiques de bases pour obtenir les familles de fragments sont également utilisées.
1.2.4 PCR : REACTION DE POLYMERISATION EN CHAINE
Lorsque l'on possède des informations (même partielles)
sur
la séquence d'un fragment d'ADN, on peut utiliser les
propriétés de l'ADN polymérase, in vitro,
pour
amplifier à l'infini ce fragment, sans faire appel à un
clonage
in vivo.
Ces dernières années, l'utilisation de la réaction
de
polymérisation en chaîne ou PCR (polymerase chain
reaction)
a révolutionné les stratégies de la
génétique
moléculaire et trouvé de très nombreuses
applications.
Le principe est illustré dans la figure suivante.
A partir de la séquence de fragments d'ADN, on va construire
deux
amorces capables de s'hybrider dans la
région
3' de chaque brin de l'ADN à amplifier. Chaque amorce sera
ensuite
allongée par une ADN polymérase.
Cette étape conduit à deux copies de la séquence
initiale.
Ces deux copies sont ensuite dénaturées et les
mêmes
amorces sont utilisées pour initier un nouveau cycle
d'élongation
conduisant à quatre copies, la répétition des
cycles
"dénaturation, hybridation des amorces,
élongation" permet une amplification exponentielle de la
séquence d'ADN située entre les deux amorces.
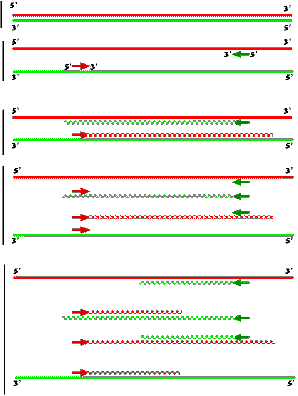 PREMIER CYCLE
PREMIER CYCLE
molécule native,
double
brin
dénaturation et hybridation des amorces
élongation des amorces par l'ADN polymérase
DEUXIEME CYCLE
dénaturation, hybridations des amorces
etc...
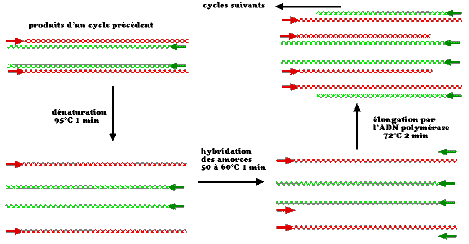 Pratiquement,
un thermocycleur permet de réaliser cette amplification sans
intervention,
pour cela, on ajoute un excès d'amorces au milieu
réactionnel
contenant l'ADN à amplifier et l'on utilise une ADN
polymérase
thermostable (extraite d'une
bactérie
se développant dans des sources chaudes : Thermus aquaticus)
résistante aux températures de dénaturation.
Pratiquement,
un thermocycleur permet de réaliser cette amplification sans
intervention,
pour cela, on ajoute un excès d'amorces au milieu
réactionnel
contenant l'ADN à amplifier et l'on utilise une ADN
polymérase
thermostable (extraite d'une
bactérie
se développant dans des sources chaudes : Thermus aquaticus)
résistante aux températures de dénaturation.
En fin d'expérience, la quantité d'ADN est telle
qu'après
électrophorèse une simple coloration permet de visualiser
la
bande correspondant au produit d'amplification.
A partir d'une très faible quantité d'ADN
génomique
au départ (5 nanogrammes par exemple) il est possible d'obtenir
un
milliard de copies d'un exemplaire unique de séquence, aucune
digestion
enzymatique préalable n'est nécessaire.
On comprend que la PCR soit rapidement devenue la méthode de
base
pour tout problème de détection de séquence
(empreintes
génétiques, repérage d'OGM ...).
Ce ne sont pas les seuls avantages de cette technique, les mêmes
amorces
peuvent, par exemple, être utilisées pour de l'ADN de
différents individus, de différentes
variétés
ou races et révéler des variations alléliques sans
clonage
biologique; elle rend de précieux services dans le domaine
du
typage génétique de la phylogénie ...).
| relire ? |
message

© Université
de TOURS - GÉNET
Document modifié le
7 janvier, 2008
![]()