|
Analyse
bioinformatique des séquences
L'approche par modélisation
Depuis des années, les bioinformaticiens se sont familiarisés progressivement avec des concepts aussi divers que le recuit simulé, les chaînes de Markov, et les statistiques bayésiennes, en l'absence d'un concept unificateur. Ce n'est que récemment qu'un cadre théorique général, plus clair et plus rigoureux, l'approche " par modélisation" ("model-driven approach" a commencé d'apparaître. Cette nouvelle reformulation est progressivement appliquée à la solutions des problèmes fondamentaux de la bioinformatique (alignement des séquences, prédiction de structure, phylogénie moléculaire, détection de gènes, etc.). Selon cette approche par modélisation, les objets (ex.Ā: séquences, structures, motifs, etc., ...) ne sont plus étudiés (alignés, classés, etc.) par comparaison directe (deux à deux ou multiple), mais à travers la construction d'un modèle qui tente, dans une première étape, d'en capturer les propriétés communes. La relation entre les objets d'étude (et/ou leur reconnaissance) est alors exprimée en référence à ce modèle optimal commun. Cette approche est résumée par le schéma suivantĀ: 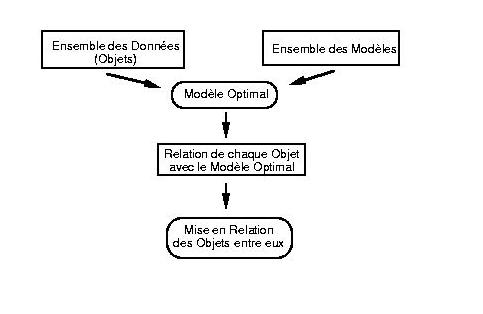 Ce schéma est naturellement lié au contexte bayésien. En effet, le modèle optimal est le plus souvent choisi comme le plus vraisemblable (le plus probable) face à l'ensemble des données disponibles (D) , c'est-à-dire comme le modèle M maximisant la probabilité conditionnelleĀ: P(M|D). En général, cette probabilité ne peut être estimée qu'à travers l'utilisation du théorème de Bayes, c'est-à-dire en utilisant la relation de proportionnalité Ā: 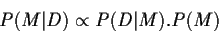 La forme (mathématique ou informatique) du modèle M est alors choisie de façon à permettre un calcul commode de la probabilité P(D|M) (la probabilité que les données observées aient été engendrées par le modèle M). De son côté, la probabilité a priori du modèle M est soit remplacée par une constante, soit estimée à partir d'hypothèses réalistes préalables. Pour l'analyse de données essentiellement linéaires (comme les séquences), les modèles probabilistes fondés sur les chaînes de Markov (simples ou cachées) se sont révélés particulièrement fructueux. Mais d'autres catégories de modèles (hypothèses évolutives, structures 3-D, etc.) sont possibles, et seront explorés dans les années à venir. On comprend aussi que les méthodes itératives et perturbatives jouent un rôle important dans ce type d'approche. En effet, le modèle optimal sera souvent choisi au terme de la convergence d'un cycle impliquant la mise en relation des objets entre eux (étape finale) dans l'estimation de la qualité des modèles intermédiaires. Les algorithmes impliqués à ce niveau sont du type "ĀExpectation-MaximizationĀ". Des techniques avancées de recherche de minimum dans des espaces de grande dimensionnalité, empruntées à des domaines scientifiques variés (par exempleĀ: "Ārecuit simuléĀ", Monte-Carlo/Metropolis, etc.) sont également nécessaires à l'étape de convergence vers les paramètres du modèle optimal. De leur côté, des algorithmes traditionnels "Ād'alignementĀ" de la bioinformatique (Needleman & Wunsch, Viterbi) continuent de jouer leur rôle dans l'étape de mise en relation (alignement) de chaque objet avec le modèle intermédiaire ou optimal. L'analyse des séquences basée sur la génération de modèles, intègre donc l'usage de concepts traditionnels de la bioinformatique, tout en les généralisant, et en leur assurant une base théorique plus rigoureuse. Le contexte probabiliste de cette nouvelle approche permet aussi d'associer les solutions proposées à une estimation de leur signification statistique. L'approche "Āpar modélisationĀ" , plus élégante, plus générale et plus rigoureuse, permet également d'espérer des progrès spectaculaires, comme l'illustre le schéma suivant:  Dans ce cas d'école, deux séquences S1 et S2 (sans la moindre position identique) sont mises en relation l'une avec l'autre par l'intermédiaire d'un "ĀmodèleĀ" M commun (par exemple une séquence ancestrale) avec lequel elles ont séparément conservé 50% d'identité. Des relations extrêmement ténues entre des séquences (des structures, ou d'autres objets d'étude) peuvent donc être mises en évidence d'une manière fiable, pour autant qu'un modèle commun puisse leur être associé. De telles idées commencent à être mises en pratique dans le contexte des méthodes d'alignement multiple, de découverte de motifs [28-31], et de classification (par exemple à l'aide de modèles structuraux sous-jacents) pour les protéines comme pour les ARNs Les mêmes principes sont aussi mis en £uvre dans le domaine de l'identification des gènes, du "ĀthreadingĀ" (mise en correspondance d'une séquence et d'un repliement), et de la phylogénie moléculaire. Résultats obtenus récemment dans les deux domaines suivantsĀ: la localisation des gènes, et la mise en évidence de motifs fonctionnels et/ou structuraux. - Exemple 1Ā: Interprétation des génomes bactériens L'identification des régions codantes d'un génome est traditionnellement vue comme un problème de recherche de "ĀsignauxĀ" de séquences, tel que la présence d'un codon initiateur, suivie d'une phase de lecture suffisamment longue (et donc de l'absence de codons STOP). Ces signaux peuvent être d'une nature plus complexe, comme certaines périodicités dans la séquence ou un biais dans l'usage des codons. La mise en évidence de ces signaux est à la base des méthodes traditionnelles [3] d'analyse des génomes. - Exemple 2Ā: l'analyse des motifs à travers leur variabilité L'arsenal expérimental de la "Āgénomique fonctionnelleĀ" est encore limité et coûteux (knock-out, transgénèse) et les méthodes d'analyse de séquences occupent une place centrale pour l'identification de la fonction des gènes, aussi bien en recherche fondamentale que pour trouver les gènes "ĀcandidatsĀ" à une application industrielle. La "ĀprédictionĀ" (en fait une classification) fonctionnelle s'effectue essentiellement par la détection d'une similarité entre une séquence nouvellement déterminée et celle d'un gène (ou de son produit) dont la fonction est connue. A un niveau plus avancé, l'alignement multiple de plusieurs séquences dotées de la même fonction est utilisé pour définir un motif consensus (ensemble de positions conservées) qui peut alors servir de "ĀsignatureĀ" fonctionnelleĀ: toute nouvelle séquence dans laquelle ce motif est détecté est alors réputée correspondre à une fonction similaire. D'une façon analogue, il est possible de définir des motifs "ĀstructurauxĀ", caractéristiques d'un certain type de repliement tridimensionnel. La définition de "ĀsignaturesĀ" permet d'étendre la classification des séquences en familles ou super-familles de fonctions identiques ou similaires, jusqu'à des cas où les similarités directes entre certaines séquences ne sont plus détectables. De nombreuses formes mathématiques différentes (matrices de position-score, profils, séquences consensus, expressions régulières, chaînes de Markov) ont été proposées pour ces signatures qui sont en fait des descripteurs (modèles) optimaux de différentes familles de séquences. Selon les méthodes courantes, un descripteur optimal d'une famille de séquence est élaboré à partir des positions les plus conservées d'un multi-alignement. Lorsque les séquences dont on dispose (ou dont on connaît la fonction) appartiennent à des organismes proches (par exemple des vertébrés), une description construite autour des positions conservées n'a qu'une faible valeur heuristique, car ces acides aminés n'ont qu'une très faible probabilité de se retrouver inchangés dans les gènes homologues d'invertébrés, de plantes, ou de micro-organismes. Or, l'application la plus importante de l'étude de ces "ĀsignaturesĀ" est la mise en évidence d'homologues distants, soit dans des systèmes-modèles pratiques (ex.Ā: la levure), soit dans des organismes pathogènes (ex.Ā: bactéries, virus). Cette nouvelle approche des signatures de séquences permet l'exploration d'une zone de faible similarité ("Ātwilight zoneĀ") auparavant inaccessible, mais néanmoins réelle. La puissance prédictive de ce type d'analyse est très fortement augmenté s'il peut être combiné à la connaissance de la structure 3-D d'une protéine, jusqu'à conduire à une prédiction fonctionnelle précise. Une fraction importante des gènes, pour l'instant classés comme "ĀorphelinsĀ" ou "ĀinconnusĀ", pourraient donc à terme rejoindre le giron d'une famille de protéines déjà décrites |
