|
Analyse
bioinformatique des séquences
Pour qualifier et quantifier la similitude entre séquences, un score est calculé. Celui-ci peut mesurer soit le rapprochement, soit l'éloignement des séquences pour refléter ce qui les sépare. Ce score repose sur un système qui permet d'attribuer un score élémentaire pour chaque position lorsque les séquences sont éditées l'une sous l'autre.

Le score élémentaire est un élément d'une matrice de scores qui rend compte de tous les états possibles en fonction de l'alphabet utilisé dans la description des séquences. Ainsi, pour les acides nucléiques, la matrice d'identité ou unitaire est principalement employée. 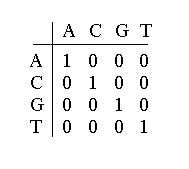
Elle rend compte de l'identité des résidus pour chacune des positions de la comparaison, on parle ainsi de bon ou de mauvais appariement ou bien de bonne ou mauvaise association. Ce critère qui permet déjà d'établir des ressemblances ne suffit pas toujours pour révéler au mieux les similitudes entre séquences. Très rapidement, on s'est aperçu qu'une insertion ou une délétion (on admettra ici le franglais) d'une ou plusieurs bases pouvait améliorer le score d'une comparaison et ainsi faire davantage ressortir les zones identiques ou très proches. Ces brèches (en anglais gap) que l'on impose aux séquences sont évidemment pénalisantes dans le calcul du score. Si l'on considère que le score donne le rapprochement entre deux séquences, on peut résumer celui-ci par l'équation suivante : où se est un score élémentaire et sp une pénalité d'insertion ou de délétion. Deux remarques s'imposent. La première est que le score est fonction de la longueur de la zone de similitude que l'on considère, c'est à dire que plus la longueur est grande, plus le score est élevé. La deuxième est que l'on peut nuancer le calcul en donnant plus ou moins d'importance aux pénalités et aux associations possibles entre résidus. Ainsi, le poids d'une insertion peut être plus ou moins fort par rapport à une mauvaise association. On voit déjà très bien ici que par le biais de ces deux éléments fondamentaux, on pourra privilégier une situation plutôt qu'une autre, c'est-à-dire avoir des comparaisons de séquences avec peu ou beaucoup d'insertions-délétions. On retrouvera bien sûr ce type d'éléments sous forme de paramètre dans les programmes de comparaison. |
