- Téléchargez le programme SIMCOAL se trouvant sur la page http://cmpg.unibe.ch/software/simcoal/, et installez le programme simcoal.exe sur votre ordinateur dans le répertoire de votre choix.
- Téléchargez le fichier dna_sta.par qui est le fichier de configuration spécifiant les paramètres des simulations à effectuer.
- Mettez le dans le répertoire ou se trouve le programme simcoal.exe.
- Consultez le contenu du fichier avec un éditeur de texte (p. ex.
notepad). Vous verrez que ce fichier va vous permettre de simuler des
échantillons
- de 20 séquences d'ADN de 300 pb
- tirés d'une population de N=5000 individus haploides
- où le taux de mutation u par séquence est de 0.0005
- Calculez rapidement (de tête) le nombre moyen de différences par paire attendu entre les séquences
- Lancez l'application simcoal.exe, entrez le nom du fichier contenant les paramètres de la simulation (dna_sta), faites faire 100 simulations
- Lancez Arlequin pour analyser les fichier simulés
- Dans Arlequn, chargez le fichier dna_sta.arb qui contient la liste de tous les fichiers générés par simcoal à partir du fichier d'entrée dna_sta.par
- Configurez les calculs à faire effectuer par Arlequin dans l'onglet
Settings. Après avoir sélectionné la ligne General settings
dans le panneau de gauche, cliquez sur le bouton Infer from
distance matrix dans le panneau de droite. Ceci oblige Arlequin à vérifier que 2
séquences ayant des noms différents soient bien différentes, de manière à calculer le nombre d'allèles dans l'échantillon.
Dans le panneau de gauche, sélectionnez la ligne Molecular diversity
indices, et cochez les cases Standard diversity indices
et Molecular diversity, ainsi que toutes les cases permettant de
calculer le paramère Theta de la population dont vous connaissez la valeur attendue.
- Estimation du paramètre q par le nombre d'allèles selon la formule (Ewens 1972)
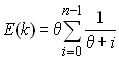
où n est la taille de l'échantillon et k le nombre d'allèle attendu.
- Estimation du paramètre q par le nombre de
sites polymorphes S
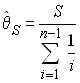
- Estimation du paramètre q par le nombre moyen de différence par paire p
E(p) = q
- Estimation du paramètre q par l'homozygotie attendue
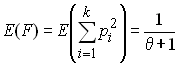 .
.
Dans l'onglet Batch file, cochez la case Theta values
Lancez les cacluls en appuyant sur le bouton Start.
Vous pouvez consulter le résumé des résultats des analyses des 100 fichiers avec Excel. Lancer Excel et droppez le fichier theta.sum généré par Arlequin. Vous verrez les estimations des valeurs de q par les 4 méthodes décrites plus haut pour chaque simulation. Calculez la moyenne des estimations et comparez les à la valeur attendue que vous avez calculé.
- Après l'avoir installée, lancez l'application Treeview.
- Glissez-collez les fichiers avec l'extension *.trees générés par simcoal.exe, et observez les généalogies simulées.