| |
Analyse
bioinformatique des séquences
6. - L'analyse de séquences nucléiques
6.4 - Les différents types de motifs [suite]
Les algorithmes exploitant des tables de fréquences
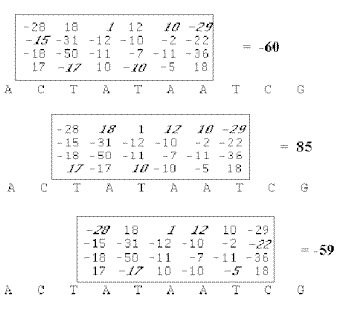
Lorsqu'un motif nucléique est défini sous forme de table de fréquences ou de probabilités, on calcule pour chaque fragment de la séquence à analyser un score. Celui-ci est déterminé en sommant les valeurs trouvées dans la table selon les bases rencontrées dans le fragment étudié et les positions considérées (Stormo,1990). Il existe en fait une correspondance entre ce score et la probabilité de trouver le motif recherché à la position déterminée par le fragment. Plus le score est élevé, plus le segment analysé a des chances de correspondre au motif recherché. Une estimation de la signification du score peut être faite en considérant les valeurs maximales et minimales théoriques données par la table et les valeurs maximales et minimales observées sur la séquence.
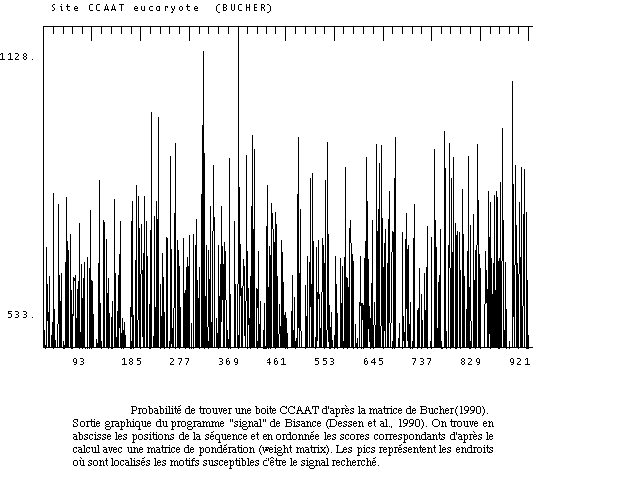
Une visualisation graphique des résultats est souvent très représentative des potentialités qu'il existe de trouver un motif le long d'une séquence.
En fait, l'intérêt principal de cette méthode réside dans la possibilité de prendre en compte une certaine similitude par rapport à un motif consensus. La plupart des logiciels possédant un ensemble de méthodes d'analyse de séquences proposent ce genre de programmes pour rechercher différents signaux nucléiques sur une séquence. Nous pouvons citer par exemple le programme MATRIX SEARCH (Chen et al, 1995) qui détermine des scores sur la séquence analysée en sommant des valeurs logarithmiques calculées à partir d'une matrice de pondération, du nombre de séquences utilisées pour établir la matrice, de la longueur du motif recherché et de la fréquence génomique des bases.
|