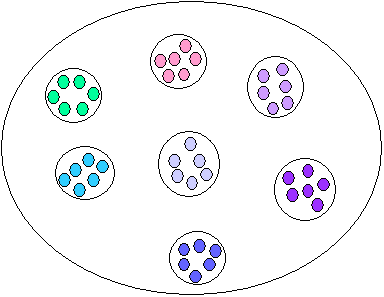
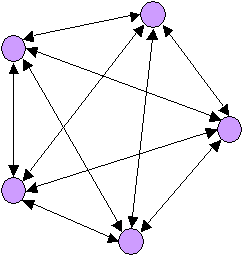
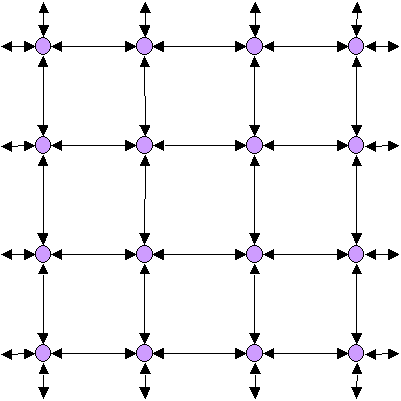
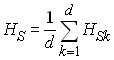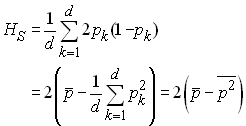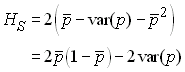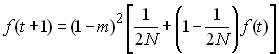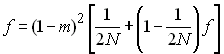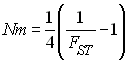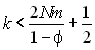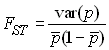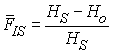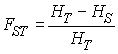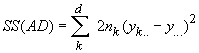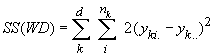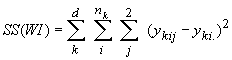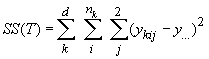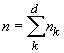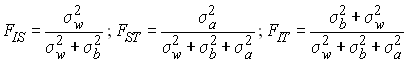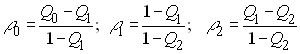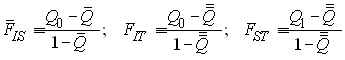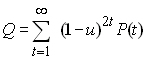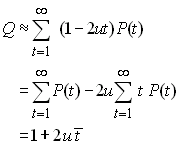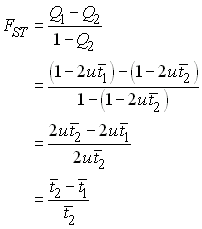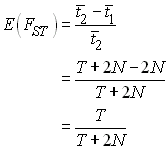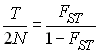Population hiérarchiquement subdivisée
| Cours I | Cours II | Cours III | Cours IV | Cours V | Cours VI |
|
Introduction |
Fréquences alléliques Dérive génétique |
Introduction à la coalescence |
Changements démographiques |
Populations subdivisées |
Sélection Tests de neutralité |
| TP I | TP II | TP III | TP IV | TP V | TP VI |
Cours V |
Population subdivisée - Modèles de migration |
Les populations naturelles ne peuvent pas toujours être considérées comme des populations panmictiques où les gamètes s'unissent au hasard. On a déjà vu un effet de l'écart à la panmixie qui est le fait que des individus apparentés s'unissent pour avoir des descendants: la consanguinité. Sa conséquence est que les deux gènes à l'intérieur d'un individu sont plus corrélés que deux gènes pris au hasard dans la population ou entre deux individus. On peut donc considérer qu'un premier niveau de subdivision est le compartiment formé par l'individu diploide qui contient deux copies d'un certain gène.
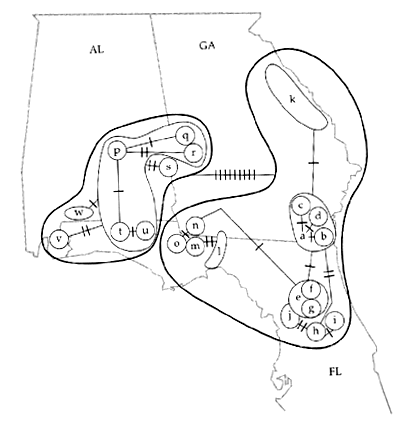
Dans les populations naturelles, les individus ne peuvent pas toujours s'unir aléatoirement sur l'ensemble de leur aire de répartition, du fait de contraintes géographique ou environnementales. Les populations seront donc presque toujours subdivisées géographiquement avec des individus qui s'unissent plus souvent avec des individus géographiquement proches qu'avec des individus éloignés. Les unités à l'intérieur desquelles les individus s'unissent le plus souvent sont appelées des dèmes. Ces dèmes peuvent être plus ou moins isolés les uns des autres, former des unités discrètes ou il peut y avoir une certaine continuité (génétique) entre dèmes voisins. Cependant, la dérive génétique va agir différemment dans chaque dème, et les dèmes vont progressivement se différencier les uns des autres.
Ces dèmes peuvent être eux-mêmes organisés en groupes dont les individus de différents s'unissent occasionnellement. On peut ainsi imaginer une espèce comme une poupée russe ayant différents degrés de subdivisions imbriqués les uns dans les autres.Donc pour la génétique des populations, une espèce est une grande population subdivisée.
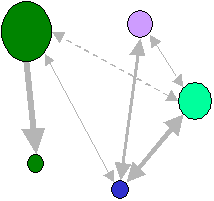
Population hiérarchiquement subdivisée
Ces dèmes peuvent maintenir des contacts entre eux par l'échange de gènes véhiculés par des migrants. La migration va donc maintenir un certain niveau de cohérence génétique entre ces dèmes.
1.1 Effet Wahlund: Excès d'homozygotes dans les populations subdivisées
On n'a pas toujours conscience que la population que l'on étudie est subdivisée. Cependant la non prise en compte de cette subdivision a comme effet un écart à l'équilibre de Hardy-Weinberg qui se traduit par un excès du nombre d'homozygotes observés par rapport aux fréquences alléliques calculées.
Pour un locus à 2 allèles qui ont des fréquences p et (1-p), la proportion d'hétérozygotes HS observés sur l'ensemble des subdivisions peut être exprimé simplement comme l'hétérozygoite moyenne
Si on a l'équilibre de Hardy-Weinberg à l'intérieur de chaque dème, alors HSk = 2 pk(1-pk) et on a donc
En se souvenant que var(X)=E(X2)-E(X)2, on a
On voit donc que la fréquence observée des hétérozygotes dans l'ensemble des subdivisions est égale à la fréquence attendue sous l'équilibre de Hardy-Weiberg en considérant la fréquence moyenne p sur l'ensemble des d dèmes moins deux fois la variance des fréquences alléliques sur l'ensemble des dèmes. En ignorant la présence de subdivisions, on va observer un déficit d'hétérozygotes, et donc un excès d'homozygotes par rapport aux fréquencs alléliques observées. Cet effet Wahlund est une conséquence directe de la présence de subdivisions à l'intérieur d'une population, et il sera d'autant plus prononcé que les populations sont plus différenciées.
Conséquence: Lorsque l'on mesure un excès significatif d'homozygotes, on peut légitimement se demander si la population étudiée n'est pas subdivisée ou amalgamé, c'est à dire qu'elle le fruit d'un rassemblement d'individus provenant de populations en équilibre de Hardy-Weinberg mais qui présentent des différences de fréquences alléliques.
Nous allons maintenant passer en revue différents modèles de population subdivisée, dontles dèmes échangent des migrants.
Dans le modèle de populations (dèmes) arrangés en îles indépendantes, comme dans un archipel. On considère que les d dèmes peuvent échanger des migrants avec tous les autres dèmes, habituellement avec la même probabilité m/(d-1). Les dèmes sont donc interconnectés par des flux de gènes identiques. Ce modèle n'est guère vraisemblable dans les populations naturelles, mais il conduit à un traitement mathématique très simple.
Modèle de 5 populations échangeant toutes une même
proportion m/4 de migrants par générationsLe processus de migration va donc interférer avec le processus de dérive à l'intérieur des dèmes. Du fait des migrations, le processus de dérive génétique ne sera plus indépendant dans chaque population, et de nouveaux allèles pourront être introduits dans des dèmes où un autre allèle s'était fixé. Cela va aussi affecter la probabilité d'identité par ascendance. Ainsi la variation du coefficient de consanguinité entre générations sera donnée par
A l'équilibre, où la perte d'allèle par dérive est compensée par l'introduction de nouveaux allèles par migration, f (t+1) = f (t) =f et
En résolvant pour f on arrive à
ce qui montre que si m>0, l'homozygotie attendue à l'intérieur de chaque dème ne tend plus vers 1, comme dans une population de taille finie, et donc on ne va plus obligatoirement fixer d'allèles.
On a vu précédemment que f pouvait être considéré comme un coefficient de corrélation. Dans notre cas, il exprime la corrélation entre 2 gènes tirés de la même subdivision (dème) par rapport à 2 gènes tirés au hasard dans la population totale. On note cette corrélation par FST, et donc l'espérance de la valeur de FST dans un modèle en île est donné par:
Le produit Nm est important car il représente le nombre absolu de migrants arrivant dans chaque dème par génération. En effet, m représente la probabilité pour un gène donné de migrer dans un dème, mais aussi la probabilité pour un gène donné qu'il soit un nouvel immigrant. A l'équilibre et en admettant que la taille de dèmes soit constante au cours du temps, le produit Nm représente donc bien la fraction attendue des gènes d'un dème qui sont de nouveaux immigrants.
L'homozygotie attendue à l'intérieur des dèmes diminue rapidement avec Nm
Nm
E( FST) 0.25 (un migrant toutes les 4 générations) 0.50 0.5 (un migrant toutes les 2 générations) 0.33 1 (un migrant par génération) 0.20 2 (deux migrants par génération) 0.11 La valeur de Nm est importante pour savoir comment vont évoluer un ensemble de populations. Moran (1962) a montré que le taux de diminution de l'hétérozygotie dans une population structurée en île et comprenant d dèmes était à peu près comparable à celui d'une population non-structurée de taille Nd si Nm >> 1. Ce résultat ne concerne que la variation du taux d'hétérozygotie. On a abusivement interprété ce résultat en disant qu'une population subdivisée où Nm est > 1 se comportait comme une seule grande population panmictique. Cela est faux pour divers aspect de la diversité génétique. C'est rassurant car on peut donc parvenir à distinguer différentes structures génétique par l'observation de la diversité génétique à l'intérieur et entre les dèmes.
On utilise souvent la relation entre Nm et la valeur de FST à l'équilibre pour le modèle en île pour estimer Nm. En faisant cela, on fait bien sûr l'hypothèse que les dèmes que l'on observe sont bien arrangés en île et que nous sommes à l'équilibre migration-dérive. Ainsi,
Chez l'homme, à l'échelle mondiale la valuer de FST est d'environ 0.1, ce qui donne une valeur de Nm de 2.25 . On a donc un degréd e différentiation entre les populations humaines qui est celle que l'on attendrait si toutes les populations étaient stationnaires et qu'elles échangeaient 2.25 migrants par génération en moyenne. Nous verrons plus tard comment on estime ces valeurs de FST .
L'effectif efficace d'une population subdivisée est en fait plus grande que celui d'une population panmictique (Whitlock et Barton 1996), et elle est dépend du degré de différentiation des dèmes comme
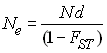
Ce résultat est dû au fait que l'effectif de chaque dème est fixe, et donc que les gènes d'individus d'un certain dème vont obligatoirement avoir des représentants à la génération suivante, et vont donc contribuer à diminuer la variance du nombre de descendants par rapport à une population panmictique. On s'attendra donc à maintenir plus de diversité génétique dans une population subdivisée que dans une population panmictique de même taille de recensement.
Kimura (1953) a introduit des modèles de migration entre dèmes plus réalistes que le modèle en île. Il a en effet voulu tenir compte du fait que l'on échangeait plutôt des migrants entre dèmes géographiquement proches les uns des autres, et pas ou rarement entre dèmes éloignés. C'est donc un modèle spatialement structué. Kimura a introduit ces modèles par analogie avec les dalles permettant de circuler dans les jardins japonais. Il a donc qualifé ces modèles en anglais de steping-stone models.
Migration en treillis en 1 dimension
Dans ce cas, les migrations se font selon un axe principal, le nombre de populations pouvant être arbitraire. Un dème va échanger des gènes avec ses deux dèmes voisins à un taux m/2.
Migration en treillis en 2 dimensions Dans ce cas, les dèmes sont arrangées selon deux axes, et un dème échangera des gènes avec ses 4 plus proches voisins à un taux m/4.
Pour éviter des effets de bord, Maruyama (1971) a introduit un modèle en une dimension mais circulaire, et un modèle en deux dimension où les dèmes sont à la surface d'un torre.
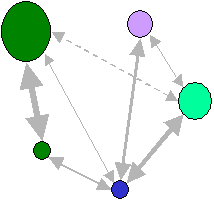
Dans ce modèle, les taux de migrations entre les dèmes sont complétements arbitraires et pas forcément symétriques. les modèles en îles et en treillis sont bien sûr des cas particuliers de ces modèles matriciels.
Les taux de migrations sont ici symétriques et proportionnels à l'épaisseur des flèches
D'une manière générale, la variabilité génétique sera plus importante dans une population avec des taux de migrations très variables entre dèmes, car il y aura des dèmes très isolés où la dérive génétique sera forte et conduira rapidment à l'élimination de certains allèles.
Les modèles de migration en treillis sont des modèles discrets d'isolement par la distance, où l'on s'attend à ce que les dèmes soient d'autant plus proches génétiquement qu'ils sont proches géographiquement. Malécot (1950) a introduit des modèles où les dèmes sont situés dans un espace continu. Les individus migrent dans un certain dème situé à une certaine distance avec une probabilité inversément proportionnelles à la distance géographique séparant ces dèmes. On tient donc compte de la distribution des distances entre le lieu de naissance des individus et de leurs descendants. D'une manière générale, que ce soit dans un espace discret ou continu, la corrélation attendue entre gènes décroit exponentiellement avec la distance.
Les modèles de méta-populations ont été introduits pour mieux coller à la réalité, et tenir compte de la nature dynamique des populations et des dèmes. Il y a en effet beaucoup de situations où les dèmes ont des tailles variables et peuvent même disparaître temprairement avant d'être crées à nouveau par de nouveaux migrants venant d'une population voisine et réoccupant le nouveau territoire. Ces propriétés sont assez typiques de petites populations, et ont donc d'importantes applications en génétique de la conservation.
Cycle d'extinction et de recolonisation dans un modèle de métapopulations
Taille de dèmes variables et taux de migration arbitraires
Un dème disparaît (extinction)
Un processus de recolonisation à partir d'un autre dème reconstitue ce dème
Le nouveau dème reprend des échanges avec les autres dèmes
D'une manière générale, des dèmes de tailles inégales sont plus différenciés que des dèmes de tailles sensiblement équivalentes, car la dérive génétique sera surtout plus forte dans les petites populations. Les extinctions de dèmes contribuent aussi à augmenter la variabilité globale de la population, en éliminant certains allèles. Enfin, les recolonisations sont souvent accompagnées d'effets fondateurs, qui entraîne de grandes perturbation de fréquences alléliques, et qui vont donc aussi contribuer à l'augmentation de la variabilité génétique entre dèmes par rapport à un ensemble de populations arrangées en îles.
Whitlock et McCauley (1990) ont montré que la différentiation génétique des dèmes sera plus grande que celle d'une population arrangée en île si
où k est le nombre moyen d'individus qui colonisent de nouveaux dèmes, N est la taille des populations qui subsistent, m est le taux de migration entre dèmes et f est la probabilité que deux gènes colonisateurs viennent du même dème source. Dans la plupart des métapopulations, cette relation est vérifiée. Si le degré de différentiation entre dèmes est plus faible que dans le cas d'une population en île cela implique que la taille efficace d'une telle population sera plus faible que dans le cas en île (voir Hedrick et Gilpin 1997 pour voir l'influence de différents facteurs sur la taille efficace, comme le nombre de subdivisions, les taux de colonisation et d'extinction. la capacité de soutien ou le nombre de fondateurs).
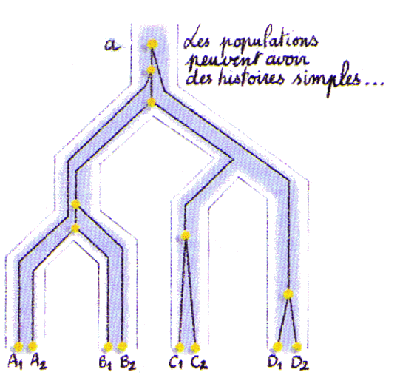
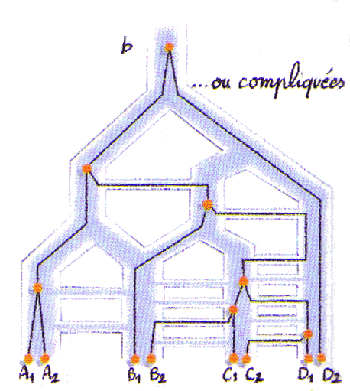
Les populations peuvent être structurées de part leur histoire. Des fissions successives de populations vont créer de nouvelles populations qui vont se différencier progressivement les unes des autres. A partir du calcul de distances génétiques entre populations, on va essayer de reconstituer ce processus de fission (voir cours d'Alicia sanchez-Mazas). Il faut toutefois noter qu'il est difficile, voire impossible, de faire la différence entre une série de fissions de populations et une population subdivisée présentant différents taux de migrations entre dèmes (Felsenstein 1982).
Admettons cependant que 4 populations se sont progressivement différenciées à partir d'une population ancestrale, comme montré ci-dessus. On va s'intéresser aux évènements de coalescences se produisant entre les différents gènes provenant de populations identiques ou différentes. Selon la taille des populations et leur temps de séparation, il se peut que la plupart des évènements de coalescence ne se produisent pas dans les populations les plus dérivées mais dans des population ancestrales. Ainsi, des gènes de différentes populations pourront très bien avoir des temps de coalescence plus petit que des gènes de la même population. Tajima (1983) a même calculé que l'on ait 95% de chance pour que les gènes de la même population coalescent avant deux gènes de populations différentes, il fallait que ces populations aient été séparées par environ 6N générations.
Conséquence: Lorsque l'on étudie la diversité moléculaire des populations d'une espèce, on ne verra à peu près jamais une correspondance parfaite entre la localisation géographique des gènes et leur position sur une phylogénie.
| Phylogénie de 56 haplotypes de restriction de l'ADN mitochondrial humain (Excoffier et al. 1992) |
|
|
Cependant il existe quand même un certain degré de cohérence géographique entre la répartition spatiale des gènes et leur position phylogénétique. Ceci est à la base des de la phylogéographie (Avise et al. 1987) qui se réfère à l'étude des processus gouvernant la distribution géographique des lignages généalogiques au niveau intra et inter spécifique. C'est véritablement l'étude de John Avise en 1979 sur le polymorphisme de l'ADN mitochondrial des gaufres de poche qui a lancé cette discipline.
Phylogénie de 87 gaufres de poche (pocket gophers) d'àprès Avise et al. (1979). Les diférents haplotypes mitochondriaux (représentés par des lettres sont reliés les uns aux autres par un réseau parcimonieux.
Lorsqu'il y a des migrations entre populations qui ont eu une histoire de fission, on s'attend à encore moins de cohérence entre localisation géographqie des gènes et relations généalogiques.
Généalogie de
gènes dans des populations
échangeant des
migrants et ayant
une histoire de
fission à partir
de populations
ancestrales.
On estime souvent le degré de subdivision d'une population au moyen de statistiques F (F-statistics) définies par Wright (1943). Ces statistiques correspondent aux corrélations de gènes pris à un certain niveau de subdivision par rapport à des gènes pris à un niveau supérieur de subdivision. On a a déjà vu que le coefficient de consanguinité f =FIS pouvait être exprimé comme la corrélation de deux gènes à l'intérieur d'un individu par rapport à deux gènes pris au hasard dans une subdivision (dans un dème). On peut définir des corrélations similaires pour des niveaux de subdivision supérieurs. On défini ainsi FST comme la corrélation de deux gènes pris dans une subdivision par rapport à deux gènes pris au hasard dans la population totale, et FIT comme la corrélation de deux gènes d'un individu par rapport à deux gènes pris au hasard dans la population totale.
On avait vu que la proportion observée d'hétérozygotes à l'intérieur d'une subdivision H est une fonction de la probabilité que les deux gènes d'un individu sont identiques par ascendance, soit
Bien que cette relation avait été dérivée dans le cas où les 2 gènes se trouvaient dans le même individu, celle-ci peut s'étendre plus généralement à deux gènes pris au hasard à n'importe quel niveau de subdivision, et par exemple à deux gènes pris au hasard dans un des dèmes de la population. Donc par extension, on a la relation
où HS est la proportion observée des hétérozygotes dans l'ensemble des subdivisions que l'on a déjà vue plus haut. S'il existe d subdivisions, HS peut aussi être considérée comme la probabilité moyenne d'être hétérozygote sur l'ensemble des d subdivisions. En remplaçant HS par la valeur trouvée par Wahlund on obtient la relation classique
qui montre que la statistique FST est également la variance observée des fréquences alléliques sur la variance attendue.
Si l'on considère un modèle où l'on part d'une seule population qui se subdivise instantanément en plusieurs dèmes tous de même taille et possédant tous les même fréquences alléliques, la variance de p sera donc nulle au dèpart du processus. Ensuite, sos l'effet de la dérive génétique, les dèmes vont peu à peu se différencier les uns des autres pour leurs fréquences alléliques. En l'absence de migrations et de mutation, un allèle ou l'autre va aller se fixer dans chaque population, et la variance de p entre les popualtion sera égale à un maximum. Dans ce cas la valeur de FST sera égale à 1. On voit donc que FST traduit en quelque sorte le degré de différentiation des dèmes dans le processus de fixation des fréquences alléliques sous l'effet de la dérive génétique. C'est pourquoi ces statistiques F sont parfois appelées des indices de fixation (fixation indexes).
D'une manière générale, les statistiques F sont reliées les unes aux autres par la relation (p.ex. Wright 1969)
La barre sur le FIS indique qu'il s'agit du coefficient de consanguinité moyen calculé sur l'ensemble des subdivisions.
Nei (1977) a montré comment calculer les statistiques F pour un nombre arbitraire d'allèles, car elles peuvent être exprimées simplement en fonction des hétérozygoties observées et attendues comme
avec Ho étant l'hétérozygotie observée dépendant des fréquences Pkii des homozygotes AiAi dans les subdivisions
HS et HT étant des hétérozygoties attendues ne dépendant pas des fréquences génotypiques, mais uniquement des fréquences alléliques dans les subdivisions pki
et bien sûr
Cockerham (1969, 1973) a montré que les corrélations entre gènes similaires aux statistiques F pouvaient être estimées par une analyse de variance des fréquences alléliques. Une telle analyse consiste à partitionner la variabilité génétique totale en divers composants de variances qui expriment la proportion de la variance totale attribués à différents niveaux de subdivision de population, soit
La variance totale étant égale à la somme des composant de variance
|
Source de variabilité |
|
|
|
Espèrance des carrés moyens |
|
Entre dèmes |
d-1 |
|
|
|
|
Entre individus à l'intérieur des dèmes |
n-d |
|
|
|
|
Entre gènes à l'intérieur des individus |
n |
|
|
|
|
Total |
2n-1 |
|
|
|
Cockerham a aussi monté que les statistiques F pouvaient être estimées par les relations
Ces estimateurs diffèrent quelque peu des estimateur de Nei à partir des hétérozygoties observées et attendues, et sont relativement moins biaisés, surtout lorsque le nombre de dèmes est la taille des échantillons est petit.
Nous avons ensuite montré (Excoffier et al. 1992) comment cette analyse de variance des fréquences alléliques pouvait être étendue pour incorporer la diversité moléculaire des gènes. On procède ainsi à une analyse de la variance moléculaire des échantillons (Analysis of MOlecular VAriance: AMOVA).
La méthode de Nei par la mesure des hétérozygoties obsevées et attendues et celle de Cockerham par l'analyse de variance fournissent des estimateurs légèrement différents. Sans trop rentrer dans les détails, cela tient au fait que les deux méthodes n'estiment pas exactement les mêmes paramétres au niveau de la population.
Si l'on définit
alors les corrélations des gènes calculées par l'analyse de variance de Cockerham sont données comme
avec r0 = FIS , r1 = FIT, et r2 = FST .
Pour les statistiques F estimées par la méthode de Nei on a les relations
où les probabilités d'identités suivantes sont définies:
Montgomery Slatkin a montré la relation entre les probabilité d'identité Q définies plus haut et les temps de coalescence (Slatkin 1991; Slatkin et Voelm, 1991). Deux gènes seront identiques si, depuis leur ancêtre commun le plus récent (MRCA), il n'y a pas eu de mutations sur aucun des 2 lignages. Supposons que cet ancêtre commun vivait il y a t générations. Dès lors, si l'on admet encore que les mutations se produisent à un rythme u par génération, la probabilité qu'aucune mutation n'est survenue pendant les 2 t générations de séparation des deux gènes est donnée par
Mais bien sûr on ne connait pas ce temps de coalescence, si bien que la probabilité non conditionnelle d'identité et obtenu en considérant tous les temps de coalescence possibles:
où P(t) est la probabilité que deux gènes coalescent au temps t et qui suit une loi géométrique comme nous l'avons vu précédemement. Si u est petit, on a la relation approximative
où ![]() est
simplement le temps de coalescence moyen de deux gènes.
est
simplement le temps de coalescence moyen de deux gènes.
On peut utiliser cette relation pour reformuler n'importe quelle statistique F en fonction de temps de coalescences moyens. Ainsi, la statistique FST estimée par l'analyse de variance devient
où ![]() et
et
![]() sont
respectivement les temps de coalescence moyens de deux gènes tirés du même
dème et deux gènes tirés de dèmes différents.
sont
respectivement les temps de coalescence moyens de deux gènes tirés du même
dème et deux gènes tirés de dèmes différents.
Ces reformulations en termes de temps de coalescence moyens permettent d'obtenir facilement les valeurs attendues des statistiques F dans différents modèles de subdivision (Slatkin 1991; Rousset 1996, 1997, 2000).
Exemple: FST dans un modèle de pure fission avec tailles de population constantes
Slatkin (1995) a considéré un modèle de populations subdivisées sans
migrations mais avec des fissions historiques. On supose qu'il y a T
générations, un ensemble de dèmes ont divergés les uns des autres et sont
restés séparés depuis cette période. Il suffit de trouver les expressions
pour ![]() et
et
![]() pour
ce modèle démographique. On a déjà vu que
pour
ce modèle démographique. On a déjà vu que ![]() = 2N pour les populations diploides et
= 2N pour les populations diploides et ![]() = N pour les populations haploides de taille N.
= N pour les populations haploides de taille N.
Maintenant, considérons des gènes de dèmes différents. On sait que les
dèmes sont restés séparés pendant T générations. Donc il n'y a pas
pu y avoir de coalescence pendant cette période. Ensuite, les lignages se sont
tous trouvés dans la population ancestrale de taille N. A partir de cet
instant, le temps moyen de coalescence de 2 lignages était à nouveau de 2N
générations (pour des population diploides. Donc, ![]() = T + 2N, si bien que
= T + 2N, si bien que
Dans ce cas, on peut estimer le temps de divergence entre les dèmes à partir du FST mesuré comme
On notera que ce temps est relatif à la taille des dèmes et de la population ancestrale, et qu'il ne dépend pas du nomre de dèmes dans la subdivision. On peut donc utiliser cet estimateur comme une mesure de distance génétique entre 2 populations.
Ce TP est accessible sur gmdp_tp5.htm
Laurent Excoffier : Dernier update : mardi, 30 mai 2006 11:18