
L'analyse du transcriptome consiste à mesurer simultanément l'expression de tous les gènes d'un génome. Elle est utilisable dans tous les problèmes où l'on suivait classiquement l'expression de quelques gènes (comparaison de plusieurs génotypes ou de plusieurs traitements, cinétique, etc.).
![]() Bibliographie
sur des exemples de variation d'expression
Bibliographie
sur des exemples de variation d'expression ![]()
La technologie qui prédomine est basée sur les puces à ADN. Une puce à ADN est constituée d'un support physique (le plus souvent une lame de verre) sur lequel sont déposées des molécules d'ADN correspondant à de petits fragments du génome (jusqu'à 40 000 dépôts différents par puce en 2007). On recouvre la puce de la solution contenant la population d'ARN à étudier. Les ARN s'hybrident sur les fragments d'ADN complémentaires. La quantité d'ARN fixée reflète la concentration de cet ARN dans la solution. Il peut exister des biais systématiques dus à d'autres facteurs, tels que l'affinité des séquences ou l'efficacité du marquage.
Pour des raisons pratiques, on utilise des ADNc plutôt que directement les ARN. Les ADNc sont marqués par un nucléotide radioactif ou un fluorochrome. Il est possible d'étudier simultanément plusieurs populations d'ADNc sur une même puce en utilisant des fluorochromes différents. La meilleure façon d'utiliser cette possibilité est de marquer de l'ADN génomique avec un fluorochrome, toujours le même. On obtient ainsi une référence stable au cours des années qui permet de mettre toutes les puces à la même échelle, quelle que soit leur origine.
Un scanner mesure l'intensité du signal émis par l'ADNc hybridé au niveau de chaque dépôt. Parmi les valeurs que proposent les logiciels pour cette intensité, la plus fiable est la médiane de l'intensité des pixels car elle est moins sensible aux défauts de l'image (pixels sur-brillants par exemple).
Les puces comportent généralement plusieurs dépôts identiques pour chaque gène. Cela simplifie le travail lorsqu'il faut repérer les aberrations dans la lecture des intensités puisqu'il suffit d'examiner les cas où les valeurs diffèrent beaucoup d'un dépôt à l'autre. Il s'agit le plus souvent d'un défaut physique sur la puce et il est facile d'éliminer la valeur aberrante. Dans le doute, on conserve la médiane des différentes mesures.
![]() Pour
en savoir plus sur les puces
Pour
en savoir plus sur les puces ![]() Bibliographie
sur les puces
Bibliographie
sur les puces ![]()
On appellera :
• expérience l'ensemble du tableau de chiffres à analyser,
• facteur un paramètre de l'expérience (un facteur de croissance, le jour de l'expérience, etc.),
• état du facteur une des valeurs qu'il peut prendre (présence ou absence du facteur de croissance, jour A, B ou C, etc.),
• condition expérimentale une combinaison particulière des états des facteurs. Une condition expérimentale correspond à une colonne du tableau de chiffres à analyser.
Les lignes du tableau correspondent aux gènes (ou à des objets apparentés tels que les EST). Une case du tableau contient une valeur qui représente, peu ou prou, le niveau d'expression d'un gène donné dans une condition expérimentale donnée.
Note : Tout ce qui est dit ici sur l'exploitation des expériences de transcriptome peut être appliqué, mutatis mutandis, aux expériences sur le protéome et le métabolome.
L'analyse du transcriptome apporte des informations statistiques qui ne prennent un sens qu'au bout d'un nombre suffisant de répétitions.
Dans une expérience de transcriptome, il est courant de constater qu'entre deux conditions expérimentales, le niveau d'expression varie notablement pour au moins 10 % des gènes. La liste est trop longue pour être exploitable concrètement. Il est nécessaire d'en extraire les gènes pertinents. Le travail est grandement facilité par une organisation adéquate des expériences. Seul un plan d'expérience bien pensé peut restreindre efficacement la liste des gènes candidats.
Cette situation permet de tirer le maximum d'informations de l'expérience pour un travail minimal. C'est un idéal dont il faut s'approcher autant que faire se peut.
Une expérience comprend trois types de facteurs, chacun apportant une information spécifique.
Le premier facteur correspond au phénomène étudié. L'étude peut porter sur deux états ou plus (deux conditions de culture par exemple ou plusieurs prélèvements au cours d'une cinétique). La situation est idéale lorsque le passage d'un état à l'autre modifie le niveau d'expression de très peu de gènes. Ainsi, par construction, la liste des gènes candidats sera courte. Typiquement, il y a plus à apprendre de la comparaison de deux maladies apparentées que de la comparaison d'un malade et d'une personne en bonne santé. En effet, les modifications du métabolisme ne sont pas toutes caractéristiques de la maladie. Il suffit pour s'en convaincre de penser à la fièvre: c'est une réaction associée à de nombreuses maladies. Malgré tout, même en prenant des précautions, il est impossible d'éviter que les gènes qui ont une relation indirecte avec le phénomène étudié polluent la liste (les gènes impliqués dans la synthèse de métabolites précurseurs, par exemple).
Le deuxième type de facteur a pour objectif de vérifier que les observations restent vraies lorsque les paramètres biologiques varient. Retrouve-t-on les mêmes changements de niveau d'expression quand l'expérience est reproduite un autre jour ? Les gènes se comportent-ils de la même façon dans différentes lignées ? . Les gènes candidats les plus intéressants sont ceux qui répondent de la même dans tous les cas (leur comportement est reproductible). Ils sont probablement au cour du phénomène étudié puisque leur comportement n'est pas limité à un contexte particulier (génétique, physiologique.). A minima, le deuxième facteur correspond à la variabilité biologique introduite par la répétition de l'expérience au cours du temps. Il est en effet quasi-impossible de réobtenir strictement les mêmes conditions physiologiques. Mais le plan d'expérience est plus efficace quand le deuxième type de facteurs est décomposé : dates, lignées, etc.
Le troisième type de facteurs correspond aux aspects techniques (protocole de marquage des ADNc, dépôt sur la puce à ADN, etc.). Ce type de facteur peut entraîner un alourdissement des expériences (dye swap) sans apporter une information biologiquement pertinente. Les artefacts techniques ne sont pas graves en soi puisque l'analyse porte sur les changements de niveau et qu'elle n'est que semi-quantitative. Les protocoles sont devenus très reproductibles et il suffit de s'en tenir à un, tout en ayant conscience qu'il présente des biais systématiques.
![]() Pour en savoir plus sur le Dye Swap
Pour en savoir plus sur le Dye Swap
![]() Bibliographie
sur la comparaison des différents protocoles
Bibliographie
sur la comparaison des différents protocoles ![]()
Pour chaque facteur, il existe deux états ou plus. L'expérience est complète si toutes les combinaisons d'états ont été effectivement mesurées. C'est à dire que le tableau des résultats complet contiendra N = n1× ... × nk× ... × nF colonnes si F est le nombre de facteur et nk le nombre de conditions pour le facteur k. La configuration typique d'une expérience complète est représentée Figure 1. Dans cet exemple, elle comporte seize colonnes.
Il est important de noter qu'une répétition biologique implique que toute la procédure expérimentale est refaite en entier à chaque fois. Dans l'exemple de la Figure 1, ceci veut dire que les deux conditions de milieu et les deux lignées ont été testées le jour A et le jour B et qu'il n'y a qu'un seul expérimentateur. Faire deux fois la lignée 1 le jour A et deux fois la lignée 2 le jour B est un non-sens car il ne sera pas possible de séparer l'effet du jour et l'effet de la lignée.
Deux échantillons sont homogènes lorsque le nombre d'individus dans chaque échantillon est suffisant pour gommer les variabilités individuelles. De la sorte, les conclusions sont valables pour l'ensemble de la population. L'échantillon est représentatif. Lorsque des raisons techniques conduisent à préparer les extraits individu par individu, on peut faire la moyenne des mesures individuelles ou mélanger les extraits avant de procéder au marquage. Les deux façons de faire donnent le même résultat. La seconde est plus économique
Chercher les gènes dont l'expression dépend de deux facteurs à la fois est une question trop vague pour qu'on puisse avoir une réponse satisfaisante. En effet, la formulation la plus générale d'une interaction revient à dire que la combinaison des états de deux facteurs donne des résultats imprévisibles. Cette définition recouvre le cas où les observations ne sont pas reproductibles !
Une étude de ce type est inéluctablement lourde. En plus des deux facteurs étudiés, il faut introduire au minimum un troisième facteur (répétition biologique.) pour être sûr que les observations sont reproductibles et biologiquement significatives.
C'est typiquement le cas des études cliniques car elles dépendent beaucoup du hasard des recrutements à l'hôpital. L'objectif du chercheur est de s'approcher autant que possible du plan d'expérience décrit ci-dessus.
Dans le cas le plus simple, les malades peuvent être regroupés de façon à former des catégories homogènes. On retombe alors dans le cas précédent en utilisant la moyenne des mesures individuelles pour chaque condition expérimentale. Les essais multicentriques remplacent la répétition biologique. Ils permettent d'éliminer les gènes dont l'expression serait propre à un fond génétique ou socioéconomique particulier.
Le problème est radicalement différent lorsque l'étude a pour objectif de découvrir des sous-types de la maladie. Dans ce cas, les malades ne peuvent pas être regroupés dans des catégories homogènes définies a priori . L'analyse statistique va chercher tout à la fois à regrouper les malades en un petit nombre de catégories et à lister les gènes dont l'expression est caractéristique de chaque catégorie. Le problème est qu'une telle analyse donne toujours un résultat. Elle aboutit inéluctablement à une liste de gènes potentiellement caractéristiques de plusieurs catégories de malades. L'étape clé va être la validation sur d'autres personnes afin de s'assurer de la fiabilité de la liste et de la réalité des catégories. La validation nécessite des effectifs importants. Il est prudent de compter cinq à dix fois plus de personnes par catégories de malades que de gènes candidats.
Il est aisé d'obtenir des gènes « diagnostiques » qui donnent d'excellents résultats au cours de la validation si les échantillons sont trop petits. Mais dans ce cas la fiabilité des résultats est illusoire et la recherche qui en découle risque fort d'être orientée sur de fausses pistes.
L'analyse du transcriptome apporte une information partielle. Elle donne une image des changements de niveau d'expression entre deux états. Mais la relation causale entre le changement d'état et le changement de niveau d'expression est indirecte pour la plupart des gènes. En fait, les changements d'état physiologique sont souvent déclenchés par l'expression transitoire d'un gène ou de quelques gènes. C'est un instant qui a peu de chance d'être saisi au cours d'une analyse de transcriptome. Identifier le ou les gènes responsables du phénomène étudié nécessite des informations complémentaires.
Une solution est d'étudier la cinétique du phénomène en mesurant l'expression des gènes à des différents temps à condition de multiplier les points de mesure pour déceler les évènements transitoires. Cette approche conduit à des expériences très lourdes car il est indispensable de répéter la cinétique plusieurs fois pour être sûr que les changements de niveau d'expression ont une réalité biologique.
Les informations provenant des séquences (motifs fonctionnels, ontologies.) et des bases de données métaboliques sont aussi très utiles. Il faut que la fonction probable des gènes au vu de leur séquence soit compatible avec le rôle suggéré par l'analyse du transcriptome.
![]() Bibliographie
sur le recoupement des sources d'informations
Bibliographie
sur le recoupement des sources d'informations ![]()
Que le marquage soit radioactif ou fluorescent, la lecture des puces à ADN fait appel à des techniques bien connues. Il n'empêche que c'est une importante source d'erreur car la dynamique du signal est élevée, l'intensité du marquage difficile à contrôler et les signaux faibles en partie cachés par le bruit de fond.
Il est fréquent que l'étendue des intensités déborde le domaine où le système de lecture a une réponse linéaire. Dans ce cas, la mesure est faussée pour les valeurs les plus faibles ou les plus fortes. Ceci conduit à des aberrations caractéristiques sur les graphiques où l'on compare les mesures faites dans deux conditions différentes (Figure 2) : le nuage des gènes est courbé et peut même présenter des stries pour les faibles intensités. En revanche, le nuage forme un cigare droit et homogène lorsque l'acquisition du signal est correcte (voir ci-dessous le chapitre Méthodes graphiques exploratoires pour une présentation détaillée du nuage des gènes).
Les erreurs de lecture peuvent être plus ou moins bien corrigées par un traitement statistique. De nombreuses méthodes permettent notamment de redresser le nuage. Ce n'est qu'un pis-aller qui ne remplace jamais une mesure correcte. La bonne solution est de prévoir sur la puce à ADN des dépôts correspondant à une gamme de concentration pour régler le système de lecture avant de procéder aux mesures. Il faut recourir à deux lectures avec des réglages différents dans le cas où la dynamique du signal excède le domaine de linéarité du système de lecture.
![]() Bibliographie
sur l'acquisition du signal
Bibliographie
sur l'acquisition du signal
Le support de la puce à ADN émet un bruit de fond dû en partie à une hybridation non spécifique. Il semble logique de supprimer ce bruit. Le problème est complexe et les options proposées par les logiciels de traitement d'image souvent simplistes. Il n'est pas rare qu'elles dégradent la qualité des mesures au lieu de l'améliorer. La correction est efficace lorsque la différence entre les deux dépôts d'un même gène est plus faible en moyenne après la correction.
En réalité le bruit de fond affecte peu l'analyse des résultats puisque celle-ci revient à examiner la position des gènes les uns par rapport aux autres dans le nuage. Sous l'effet du bruit de fond, les gènes bougent un peu, mais pas au point de bouleverser le nuage. L'effet n'est sensible que pour les plus bas niveaux d'expression, dans un cas où de toute façon les mesures sont très imprécises.
Les sources de variabilités incontrôlées sont nombreuses et une préparation d'ARNm a peu de chance de donner les mêmes résultats si elle est testée sur deux puces à ADN : il n'y a pas de raisons pour que le marquage soit exactement le même d'une fois sur l'autre ou que le système de lecture soit réglé exactement de la même façon.
Ce sont des biais qui affectent tous les gènes de la même façon et le problème est résolu en donnant la même moyenne et la même variance à toutes les conditions expérimentales (c'est-à-dire toutes les colonnes dans le tableau de résultats). D'un point de vue statistique, cette opération revient à leur donner le même poids dans les analyses ultérieures. D'un point de vue biologique, c'est faire l'hypothèse que la quantité totale d'ARNm dans les cellules est constante dans l'expérience. L'hypothèse est solide pour une puce génomique car l'expression de la plupart des gènes ne change pas dans une expérience. Elle est à vérifier au cas par cas avec les puces spécialisées contenant peu de gènes. Certains auteurs préfèrent utiliser une estimation robuste de la moyenne et de la variance basée sur les quartiles (Encart 1). Cette façon de faire est moins sensible aux valeurs extrêmes (anormalement basses ou anormalement élevées), et parfois mieux adaptéeà la distribution des valeurs d'expression.
La distribution du niveau d'expression des gènes est très asymétrique avec un petit nombre de valeurs élevées. C'est une source de problèmes car de nombreuses méthodes statistiques supposent implicitement une distribution gaussienne. L'asymétrie est fortement diminuée si les données brutes sont remplacées par leur logarithme ou par leur racine cinquième. La transformation logarithmique est la plus utilisée. Après transformation, les méthodes statistiques peuvent être utilisées en toute confiance.
La transformation logarithmique présente l'inconvénient d'amplifier les écarts des petites valeurs : après le passage au logarithme, 0,1 et 1 deviennent aussi éloignés que 100 et 1000. Le nuage des gènes s'évase considérablement pour prendre une forme en trompette (Figure 3). Cette figure très fréquente dans les publications est un pur artefact dû à la transformation logarithmique de valeurs proches de zéro. Nota bene, les valeurs proches de zéro résultent habituellement de la soustraction du bruit de fond.
Des traitements mathématiques permettent de limiter l'ampleur de cette déformation. Le problème peut être évité en ajoutant une constante de sorte que la valeur la plus faible soit aux environs de la centaine.
Bibliographie sur la
normalisation ![]()
![]()
Les dépôts correspondant à chaque gène ne sont pas toujours reproductibles d'une puce à l'autre (à cause, par exemple, d'une concentration variable de la solution d'ADN qui est déposée). Il est possible de corriger ce biais et de mettre toutes les puces à la même échelle à condition de les hybrider toutes avec la même référence.
La meilleure référence est l'ADN génomique marqué toujours avec le même fluorochrome car elle est parfaitement stable dans le temps. L'utilisation de l'ADN génomique allège considérablement les plans d'expérience en supprimant notamment le dye swap et permet de mettre à la même échelle des puces qui ont été fabriquées à des années d'écart.
L'expérience montre que la PCR quantitative donne les mêmes résultats que les puces à ADN lorsqu'on utilise les mêmes sondes. Une expérience réalisée avec des PCR quantitatives est donc l'équivalent d'une analyse de transcriptome menée avec une puce à ADN comportant peu de gènes.
Par conséquent, toutes les méthodes d'analyse développées pour les puces à ADN s'appliquent à la PCR quantitative, et en particulier la nécessité d'une transformation logarithmique (pour rendre les distributions gaussiennes) suivie d'une transformation linéaire pour amener la moyenne à zéro et la variance à un pour chaque condition expérimentale. Dans le cas de la PCR quantitative, ceci revient à appliquer la transformation linéaire directement sur les Delta Ct (Ct = nombre de cycles d'amplification à partir duquel la fluorescence est détectée ; Delta Ct = Ct gène étudié - Ct gène référence).
![]() Bibliographie
sur la PCR quantitative
Bibliographie
sur la PCR quantitative
Les valeurs manquantes posent un problème car la plupart des analyses nécessitent un tableau de chiffres complet. Les valeurs manquantes ont deux origines : (i) un défaut rend la mesure impossible pour un gène sur une puce à ADN ; (ii) la mesure est éliminée car elle n'est pas notablement supérieure au bruit de fond.
Eliminer un gène lorsqu'il lui manque une valeur dans une colonne n'est pas une solution car le problème va se poser de nouveau pour d'autres gènes dans d'autres colonnes si le nombre de conditions expérimentales augmente. On considère généralement qu'il vaut mieux remplacer la valeur manquante par une valeur estimée à partir des mesures faites dans d'autres conditions expérimentales.
En revanche, supprimer une valeur bruitée, mais réelle, pour la remplacer par une valeur estimée est un contre-sens. On perd l'information qui était plus ou moins noyée dans le bruit de fond sans rien gagner en contre-partie. C'est pourtant ce que l'on fait lorsqu'on élimine les valeurs voisines du bruit de fond.
Plusieurs techniques, dont la régression linéaire, permettent de calculer les valeurs estimées. Elles n'ont pas toutes la même précision. On peut estimer empiriquement la qualité d'une méthode en supprimant quelques valeurs dans le tableau de départ puis en comparant les valeurs prédites aux valeurs initiales.
Bibliographie sur le traitement
des valeurs manquantes ![]()
![]()
On fait appel aux statistiques pour répondre à la question : les différences d'expression observées sont-elles bien réelles ? La réponse est indirecte, les statistiques donnent la probabilité pour qu'on ait affaire à un faux-positif (la p-value ). Un faux-positif correspond au cas d'un gène où la différence observée dépasse par hasard un seuil fixé à l'avance. « Par hasard » signifie qu'en général on ne retrouverait pas une différence aussi grande si l'expérience était répétée.
Comme une expérience de transcriptome porte sur des milliers de gènes simultanément, l'analyse statistique est utilisée pour évaluer le nombre probable de faux-positifs au-delà d'un seuil donné : 40 gènes ont une p-value inférieure ou égale à 1 % par hasard si l'expérience porte sur 4 000 gènes alors que c'est le cas de 400 gènes si l'expérience porte sur 40 000 gènes.
L'estimation du nombre de faux positifs n'est qu'une première étape dans le raisonnement lorsqu'on fixe le seuil qui va définir l'ensemble des gènes à étudier. En effet, on trouve au-delà du seuil à la fois des faux positifs et des gènes pour lesquels la différence observée est bien réelle (c'est-à-dire qu'on retrouverait ces gènes dans une autre expérience).
L'information clé est la proportion de faux positifs dans l'ensemble des gènes sélectionnés car elle mesure le risque de se lancer dans une fausse piste si on décide de travailler sur un gène pris dans cet ensemble. C'est le False Discovery Rate (FDR). Habituellement le seuil est fixé de sorte qu'il n'y ait pas plus de 5 % de faux-positifs dans le lot de gènes sélectionnés (FDR = 5 %).
Par exemple, prenons une expérience portant sur 4 000 gènes dont 80 gènes ont une p-value inférieure ou égale à 0,1 % (p=0,001) qui sont les gènes retenus comme potentiellement intéressants. Sur 4000 gènes au départ, il y a environ 4 faux positifs (4000 × 0,001). Sur les 80 gènes retenus, il y en a donc environ que 4 qui par hasard présentent une p-value inférieure ou égale à 0,001. Plus précisément, le pourcentage de faux positifs (FDR) est de 4 / 80, soit 5 % des gènes sélectionnés.
La littérature fait parfois référence à la correction de Bonferroni. Cette correction n'est pas pertinente pour l'analyse du transcriptome car elle est exagérément restrictive.
Le critère numérique utilisé dans un test statistique est toujours le rapport des écarts observés pour le facteur intéressant (le signal) sur ceux qui sont dus à l'ensemble des causes qu'on néglige (le bruit). Les tests statistiques se distinguent par la façon de définir le bruit et par la loi utilisée pour estimer la probabilité des faux-positifs.
Plusieurs grandeurs sont utilisées simultanément pour décider si l'expression d'un gène varie de façon significative pour le facteur étudié :
• V1, la variance de l'ensemble des observations faites sur le gène,
• V2, la variance des observations pour le facteur étudié (ou une grandeur apparentée comme l'écart entre les deux moyennes dans le cas où le facteur n'a que deux états),
• V3, la variance des observations pour les facteurs dont on souhaite soustraire l'influence.
Le bruit est égal à V1 - (V2 + V3) et le signal à V2. La possibilité de calculer le terme V3 est une spécificité de l'analyse de variance (ANOVA), elle permet de contrôler plus finement la composition du bruit. Dans l'exemple du plan d'expérience de la Figure1, V3 correspond aux changements de niveau d'expression induits par le jour, la lignée et les problèmes techniques. Et le bruit recouvre tout ce qui fait que le niveau d'expression réel diffère de la simple addition des effets du facteur étudié, du jour, de la lignée et des problèmes techniques.
La difficulté majeure est de cerner le bruit avec précision tout en l'évaluant sur suffisamment de données. L'approche la plus sûre est de répéter l'expérience un grand nombre de fois. Ce n'est pas toujours possible et le nombre d'observations est souvent inférieur à 20. Les statisticiens cherchent alors à améliorer l'estimation du bruit en travaillant sur des groupes de gènes qui présentent à peu près le même niveau de bruit. De nombreuses solutions sont possibles, aucune n'est parfaite. Généralement, les regroupements sont faits a posteriori, après une première estimation du bruit pour tous les gènes séparément. On parle d'approche bayesienne .
Les méthodes se distinguent aussi par la distribution statistique du bruit. Elles prennent soit une distribution définie a priori (le plus souvent la gaussienne) soit une distribution estimée par permutation à partir de l'échantillon (les couples valeurs observées × conditions expérimentales sont constitués au hasard).
Les différentes méthodes ne mesurent pas exactement la même chose puisqu'elles n'évaluent pas le bruit de la même façon. Il est naturel qu'elles ne donnent pas exactement les mêmes résultats (Figure 4).
Les meilleures méthodes ont des sensibilités équivalentes en moyenne, mais différentes au cas par cas. Elles apportent des informations partiellement complémentaires. Il est logique de comparer leur résultat. Par contre, il ne faut pas se limiter à la liste des gènes significatifs avec toutes les méthodes. En effet, cela reviendrait à choisir pour chaque gène la méthode la moins sensible. En d'autres termes, il faut travailler sur l'union des listes pas sur leur intersection.
Le choix du test statistique joue un rôle secondaire dans la finesse des analyses. Celle-ci dépend avant tout du plan d'expérience car c'est lui qui permet d'éliminer une grande partie du bruit (voir ci-dessus le chapitre Concevoir un plan d'expérience ).
Bibliographie sur les statistiques ![]()
![]()
Les résultats d'une expérience de transcriptome forment un tableau de K colonnes, les conditions expérimentales ou les patients, et L lignes, les gènes. Il y a habituellement plusieurs milliers de gènes et quelques dizaines de colonnes. La cellule lk contient la valeur observée pour le gène l dans la condition k (son niveau d'expression) En d'autres termes, l'expérience peut être représentée par un unique nuage de L points (un par gène) dans un espace ayant K dimensions (une par condition expérimentale). La cellule lk est la coordonnée du gène l dans la condition k et la ligne l correspond aux coordonnées du gène l dans l'espace de l'expérience.
Un seul coup d'oil donnerait une vision complète de l'expérience si nous pouvions nous représenter un objet dans un espace à K dimensions. Mais nous sommes limités à deux dimensions (trois avec des astuces graphiques) et il va falloir aborder le nuage par plans successifs. L'idée simple qui consiste à faire tous les graphiques en prenant les conditions expérimentales deux par deux n'est pas réaliste car le nombre de graphiques est généralement très grand (il y a K×(K-1) / 2 graphiques, c'est-à-dire 120 graphiques rien que pour les seize conditions expérimentales de la Figure 1). Il n'est pas possible de les analyser tous et, de toute façon, chacun d'eux ne contient qu'une toute petite partie de l'information. Il est nécessaire de regrouper astucieusement les conditions expérimentales pour aboutir à un petit nombre de graphiques réellement pertinents.
Nota bene , dans tout ce qui suit, on appelle « niveau d'expression » la valeur normalisée de l'intensité lue par le photomultiplicateur (la normalisation correspond à une transformation logarithmique suivie d'une transformation linéaire pour amener la moyenne à zéro et la variance à un pour chaque condition expérimentale).
Une représentation classique consiste à mettre en abscisse le niveau moyen d'expression dans l'ensemble de l'expérience et en ordonnée la différence des niveaux d'expression entre deux états différents (Figure 5, image du haut). Dans l'idéal, si les observations étaient parfaitement reproductibles, l'ordonnée serait égale à zéro pour les gènes dont le niveau d'expression ne varie pas dans l'expérience : ils seraient alignés sur une droite D . En réalité, le nuage des gènes ne forme pas une droite mais un cigare très allongé car les observations sont bruitées. Le bruit correspond justement à l'épaisseur du nuage.
Les gènes dont le niveau d'expression a changé sont en dehors de la droite D . Leur distance à la droite est proportionnelle à la différence des niveaux d'expression. Ils sont faciles à repérer lorsqu'ils figurent à la périphérie du nuage, c'est-à-dire quand le rapport signal sur bruit est élevé (le signal étant la distance à la droite D et le bruit l'épaisseur moyenne du nuage au même endroit).
L'image du haut de la Figure 5 est simple à construire puisqu'elle donne le même poids à toutes les conditions expérimentales. Mais ce choix qui n'est pas pertinent lorsque certaines conditions expérimentales apportent plus d'information que d'autres (par exemple des mesures faites en partant de peu d'ARN sont moins précises que les autres). La meilleure solution est de remplacer la moyenne simple par une moyenne pondérée où le poids d'une condition expérimentale est proportionné à l'information qu'elle apporte. Le calcul de la pondération optimale est possible sous certaines hypothèses.
L'analyse en composantes principales (ACP) donne la solution lorsque le niveau d'expression a une distribution gaussienne (une distribution gaussienne est totalement définie par sa variance). Les figures produites par l'ACP contiennent toute l'information car elles sont calculées de telle sorte que la variance de chacune est maximale. Elles donnent une description complète de l'expérience.
Mais en réalité la distribution du niveau d'expression est souvent différente d'une gaussienne (distribution avec des écarts à la moyenne très importants, distribution asymétrique, distribution bi-modale, etc.). Dans ce cas l'ACP ne fournit pas la solution optimale. Il est préférable de la remplacer par l'analyse en composantes indépendantes (ACI). L'ACI montre le nuage sous des angles mettant en valeur les anomalies de distribution. Elle permet de déceler des phénomènes qui ont échappé avec l'ACP.
Utiliser plusieurs méthodes graphiques est équivalent à regarder le nuage sous des angles différents. Il est imaginable de voir sous un certain angle des gènes sortir du nuage alors qu'ils paraissent noyés dans la masse sous un autre angle (Figure 5). Le seul risque que l'on court avec une méthode inadéquate est rater des gènes.
En général, seule la périphérie du nuage est exploitable visuellement. L'organisation interne est cachée par la superposition de milliers de gènes sur une même image. Il est pourtant intéressant d'identifier les gènes qui sont proches les uns des autres dans le nuage car ce sont des gènes qui ont à peu près le même profil d'expression. Les biologistes posent le plus souvent une question apparentée : quels sont les gènes impliqués dans un même processus ? Y répondre revient à chercher dans le nuage des régions où la densité de gènes est anormalement forte (en d'autres termes, cela revient à chercher les amas de gènes dans le nuage).
Il s'agit en réalité d'une question très générale qui se pose dans la plupart des disciplines scientifiques et qui a donné naissance à des myriades de méthodes de classification (on dit aussi clustering). Derrière leur apparente simplicité, les méthodes de clustering nécessitent toujours de fixer la valeur de plusieurs paramètres. C'est un exercice difficile car le jeu de valeurs qui donne de bons résultats dans certains cas peut s'avérer très médiocre sur d'autres données. Si toutes les méthodes donnent à peu près le même résultat quand le nuage est fragmenté avec des amas clairement séparés par des zones pratiquement vides, les résultats sont beaucoup moins fiables quand le nuage est homogène. Ils dépendent alors beaucoup de la méthode de classification utilisée et de la valeur des paramètres (Figure 6).
Faute de critères objectifs, il n'est pas rare alors que le biologiste retienne seulement les clusters qu'il sait interpréter. L'analyse du transcriptome ne lui sert alors qu'à confirmer ce qu'il savait déjà, ce qui n'est pas nécessairement le résultat le plus intéressant ! En d'autres termes, le clustering convient bien pour illustrer un résultat obtenu par une autre approche, mais ce n'est pas une bonne méthode pour découvrir quelques chose.
L'analyse serait facilitée s'il était possible de donner une image fidèle de la densité de gènes dans chaque région du nuage avec peu ( j) de points, puis de lister les gènes qui sont dans les régions où ces points sont proches les uns des autres. Une façon de faire est de redessiner le nuage en tirant au sort j gènes (par exemple un gène sur cent). C'est une solution naïve qui a peu de chance d'être satisfaisante. Une autre solution assez simple est le k-means . Elle vise à regrouper les points en k groupes aussi denses que possible (k est fixé par l'utilisateur). En fait, calculer la position optimale des j points pour donner une image fidèle des variations de densité au sein du nuage est un problème difficile. Les programmes ne proposent que des solutions approchées dont le détail dépend de plusieurs paramètres. Un exemple de cette approche est donné par SOM (Self Organizing Maps).
Une autre solution est d'agglomérer progressivement les points en commençant par ceux qui sont les plus proches dans le nuage. On parle alors de classification hiérarchique.
On peut dans le cas du transcriptome utilisé un artifice qui consiste à éliminer les gènes qui sont au cour du nuage avant de procéder à une classification. Cette élimination se fait après analyse statistique des données. Ainsi l'analyse ne porte que sur ceux dont le niveau d'expression a changé notablement au cours de l'expérience.
Dans tous les cas, il faut vérifier la stabilité de la classification obtenue. Pour cela on bruite les données initiales en les modifiant aléatoirement de 10 à 20 %, puis on relance la classification. Par recoupement, il est possible de repérer les gènes qui sont toujours classés ensembles.
![]() Bibliographie
sur les méthodes de clustering
Bibliographie
sur les méthodes de clustering
Le transcriptome est fréquemment utilisé pour identifier un état physiologique particulier (par exemple pour optimiser le traitement en fonction du sous-type de cancer). L'objectif est d'obtenir un diagnostic fiable basé sur la mesure du niveau d'expression de quelques gènes.
C'est un problème différent de celui traité ci-dessus. D'un point de vue géométrique, les points du nuage sont maintenant des individus. Les coordonnées d'un individu dans le nuage correspondent au niveau d'expression de ses gènes. On cherche à représenter le nuage sous un angle qui sépare le plus possible les différentes catégories d'individus (maladie 1 vs maladie 2 par exemple). En pratique, on souhaite obtenir une bonne séparation en utilisant le moins possible de gènes (ceux qui sont les plus discriminants). Il est ensuite possible de placer un nouvel individu sur la figure pour voir à quelle catégorie il se rattache.
De nombreuses méthodes traitent ce problème. Elles diffèrent notamment par la façon de tracer la frontière entre les différentes catégories d'individus (Figure 7). Dans la méthode SVM ( Support Vector Machines ou Séparatrices à Vaste Marge en français), la frontière est tracée de sorte à maximiser la largeur de la marge qui sépare les différentes catégories d'individus (image D de la Figure 7). SVM présente aussi l'avantage d'identifier les individus qui sont à la marge et qui sont, en quelque sorte, les moins typiques de leur catégorie.
![]() Pour
en savoir plus sur la méthode SVM.
Pour
en savoir plus sur la méthode SVM.
![]() Bibliographie
sur le choix des gènes
Bibliographie
sur le choix des gènes ![]()
Jusqu'ici nous avons implicitement considéré que la distance entre les gènes était le critère pertinent pour examiner leurs relations au sein du nuage. Il y a pourtant d'autres choix possibles. Par exemple si les points du nuage avaient une masse, le critère pertinent serait le carré de la distance et non pas la distance linéaire car l'attraction entre eux serait inversement proportionnelle au carré de leur distance. Dans un autre ordre d'idée, on peut imaginer que la probabilité pour que deux gènes appartiennent à la même famille fonctionnelle décroît très vite lorsque la distance entre les gènes augmente, la décroissance pouvant suivre une gaussienne, une exponentielle ou toute autre loi de probabilité.
On parle de méthode à noyau dans tous les cas où la distance linéaire est remplacée par un critère non linéaire. Cette technique augmente la puissance des méthodes d'analyse. Elle est appliquée couramment à des méthodes comme SOM et SVM. Elle peut aussi être utilisée pour l'ACP et l'ACI, qui deviennent alors des ACP et des ACI à noyau.
![]() Pour
en savoir plus sur les méthodes à noyau
Pour
en savoir plus sur les méthodes à noyau
Le plus souvent, les expériences de transcriptome sont sous-exploitées car l'expérimentateur ne s'intéresse qu'à quelques gènes ou à un facteur donné alors que les observations portent sur tous les gènes et l'ensemble des facteurs. Une exploration du nuage dans toutes les directions ouvre la voie à une exploitation beaucoup plus complète des observations. L'analyse en composantes principales et l'analyse en composantes indépendantes facilitent ce type d'approche.
Il est fréquent d'isoler ainsi des groupes de gènes dont le profil d'expression est propre à quelques patients ou quelques conditions expérimentales. La cause des changements de niveau d'expression est souvent inconnue car elle ne coïncide par avec un facteur clairement identifié dans l'expérience. Malgré tout l'observation est importante. Elle peut déboucher sur l'identification de sous-types dans une maladie ou contribuer à la découverte de réseaux de gènes.
Il est rarement possible de monter une expérience de transcriptome d'une taille suffisante pour identifier avec certitude des sous-types dans une maladie ou un réseau de gène. Cependant, on peut répondre à ces questions même si l'on ne connaît pas en détail les conditions expérimentale. C'est pourquoi la faiblesse de l'échantillon peut être compensée en exploitant les données accessibles sur le Web. Un pré-traitement statistique corrige l'hétérogénéité des données.
Vous pouvez télécharger la bibliographie complête (fichier pdf ou fichier html) en cliquant ici ou la consulter ci-dessous (lien sur tous les articles au format pdf):
Sommaire de la bibliographie
An improved physico-chemical model of hybridization
on high-density oligonucleotide microarrays
Comparison of Affymetrix GeneChip expression measures
Evaluation of methods for oligonucleotide array data
via quantitative real-time PCR..
Statistical analysis of high-density oligonucleotide
arrays: a multiplicative noise model
The high-level similarity of some disparate gene expression
measures
Adaptative quality-based clustering of gene expression
profiles
Analysing microarray data using modular regulation analysis
Cluster analysis and display of genome-wide expression
patterns
Comparisons and validation of statistical clustering
techniques for microarray gene expression data
Determination of minimum sample size and discriminatory
expression patterns in microarray data
Evolutionary algorithms for finding optimal gene sets
in microarray prediction
From co-expression to co-regulation: how many microarray
experiments do we need?
Instance-based concept learning from multiclass DNA
microarray data
Trustworthiness and metrics in visualizing similarity
of gene expression
Using repeated measurements to validate hierarchical
gene clusters
Weighted rank aggregation of cluster validation measures:
a Monte Carlo cross-entropy approach
A review of feature selection techniques in bioinformatics
Bioinformatics need to adopt statistical thinking
Comments on selected fundamental aspects of microarray
analysis
Evaluation and comparison of gene clustering methods
in microarray analysis
Evaluation of microarray data normalization procedures
using spike-in experiments
Metabolomics in systems biology
Microarray data analysis: from disarray to consolidation
and consensus
Overview of Tools for Microarray Data Analysis and Comparison
Analysis
Cross-platform
reproducibility
A methodology for global validation of microarray experiments
A study of inter-lab and inter-platform agreement of
DNA microarray data
Analysis of variance components in gene expression data
Application of a correlation correction factor in a
microarray cross-platform reproducibility study
Reproducibility of microarray data: a further analysis
of microarray quality control (MAQC) data
Three microarray platforms: an analysis of their concordance
in profiling gene expression
A framework for significance analysis of gene expression
data using dimension reduction methods
AnovArray: a set of SAS macros for the analysis of variance
of gene expression data
Blind Source Separation and the Analysis of Microarray
Data
Correspondence analysis applied to microarray data
GeneANOVA gene expression analysis of variance
Gene expression variation between mouse inbred strains
Independent Component Analysis: A Tutorial
Linear modes of gene expression determined by independent
component analysis.
Metabolite fingerprinting: detecting biological features
by independent component analysis
Novel approaches to gene expression analysis of active
polyarticular juvenile rheumatoid arthritis
Significance analysis of microarrays applied to the
ionizing radiation response
Statistical Design and the Analysis of Gene Expression
Microarray Data
Variation in tissue-specific gene expression among natural
populations
A comparative review of estimates of the proportion
unchanged genes and the false discovery rate
A note on the false discovery rate and inconsistent
comparisons between experiments.
A simple method for assessing sample sizes in microarray
experiments
Effects of dependence in high-dimensional multiple testing
problems
Empirical Bayes screening of many p-values with applications
to microarray studies
Estimating p-values in small microarray experiments
Classification of microarray data using gene networks
Enrichment or depletion of a GO category within a class
of genes: which test?
Ontological analysis of gene expression data: current
tools, limitations, and open problems
An empirical Bayes approach to inferring large-scale
gene association networks
Analyzing gene expression data in terms of gene sets:
methodological issues
Comparative evaluation of gene-set analysis methods
Pathway level analysis of gene expression using singular
value decomposition
Bayesian meta-analysis models for microarray data: a
comparative study
Can subtle changes in gene expression be consistently
detected with different microarray platforms?
Coexpression Analysis of Human Genes Across Many Microarray
Data Sets
Combining Affymetrix microarray results
Merging two gene-expression studies via cross-platform
normalization
Variation in tissue-specific gene expression among natural
populations
Ranking analysis of F-statistics for microarray data
An adaptive method for cDNA microarray normalization
Can Zipf's law be adapted to normalize microarrays?
Making sense of microarray data distributions
Normalization of single-channel DNA array data by principal
component analysis
Reuse of imputed data in microarray analysis increases
imputation efficiency
Using Generalized Procrustes Analysis (GPA) for normalization
of cDNA microarray data
Biases induced by pooling samples in microarray experiments
Effect of pooling samples on the efficiency of comparative
studies using microarrays
Pooling mRNA in microarray experiments and its effect
on power
Statistical implications of pooling RNA samples for
microarray experiments
Quality
control of microarrays
A Bayesian missing value estimation method for gene
expression profile data
A comparison of background correction methods for two-colour
microarrays
Combining signals from spotted cDNA microarrays obtained
at different scanning intensities
Comparing transformation methods for DNA microarray
data
Correcting for gene-specific dye bias in DNA microarrays
using the method of maximum likelihood
Gaussian mixture clustering and imputation of microarray
data
Microarray image analysis: background estimation using
quantile and morphological filters
Missing-value estimation using linear and non-linear
regression with Bayesian gene selection
Quality determination and the repair of poor quality
spots in array experiments
Scanning microarrays at multiple intensities enhances
discovery of differentially expressed genes
Statistical estimation of gene expression using multiple
laser scans of microarrays
Model based analysis of real-time PCR data from DNA
binding dye protocols
Statistical analysis of real-time PCR data
Statistical significance of quantitative PCR
Analyzing time series gene expression data
Difference-based clustering of short time-course microarray
data with replicates.
Fundamental patterns underlying gene expression profiles:
Simplicity from complexity
Identification of gene expression patterns using planned
linear contrasts
Permutation test for periodicity in short time series
data
A calibration method for estimating absolute expression
levels from microarray data
An analysis of the use of genomic DNA as a universal
reference in two channel DNA microarrays
An experimental evaluation of a loop versus a reference
design for two-channel microarrays
Analysis of Variance for Gene Expression Microarray
Data
Background correction for cDNA microarray images using
the TV+L1 model
Characterizing dye bias in microarray experiments
Effect of various normalization methods on Applied Biosystems
expression array system data
Evaluation of the gene-specific dye bias in cDNA microarray
experiments
Comment on Evaluation of the gene-specific dye bias
in cDNA microarray experiments
Extended analysis of benchmark datasets for Agilent
two-color microarrays
Missing channels in two-colour microarray experiments:
Combining single-channel and two-channel data
Reducing the variability in cDNA microarray image processing
by Bayesian inference
Pre-processing Agilent microarray data
![]()
Naoaki
Ono, Shingo Suzuki, Chikara Furusawa, Tomoharu Agata, Akiko Kashiwagi,
Hiroshi Shimizu and Tetsuya Yomo
Motivation: High-density DNA microarrays provide
useful tools to analyze gene expression comprehensively. However, it is
still difficult to obtain accurate expression levels from the observed
microarray data because the signal intensity is affected by complicated
factors involving probetarget hybridization, such as non-linear behavior
of hybridization, non-specific hybridization, and folding of probe and
target oligonucleotides. Various methods for microarray data analysis have
been proposed to address this problem. In our previous report, we presented
a benchmark analysis of probe-target hybridization using artificially synthesized
oligonucleotides as targets, in which the effect of non-specific hybridization
was negligible. The results showed that the preceding models explained
the behavior of probe-target hybridization only within a narrow range of
target concentrations. More accurate models are required for quantitative
expression analysis.
Results: The experiments showed that finiteness
of both probe and target molecules should be considered to explain the
hybridization behavior. In this article, we present an extension of the
Langmuir model that reproduces the experimental results consistently. In
this model, we introduced the effects of secondary structure formation,
and dissociation of the probe-target duplex during washing after hybridization.
The results will provide useful methods for the understanding and analysis
of microarray experiments.
Rafael
A. Irizarry, Zhijin Wu and Harris A. Jaffee
Motivation: In the Affymetrix GeneChip system,
preprocessing occurs before one obtains expression level measurements.
Because the number of competing preprocessing methods was large and growing
we developed a benchmark to help users identify the best method for their
application. A webtool was made available for developers to benchmark their
procedures. At the time of writing over 50 methods had been submitted.
Results: We benchmarked 31 probe set algorithms
using a U95A dataset of spike in controls. Using this dataset, we found
that background correction, one of the main steps in preprocessing, has
the largest effect on performance. In particular, background correction
appears to improve accuracy but, in general, worsen precision. The benchmark
results put this balance in perspective. Furthermore, we have improved
some of the original benchmark metrics to provide more detailed information
regarding precision and accuracy. A handful of methods stand out as providing
the best balance using spike-in data with the older U95A array, although
different experiments on more current arrays may benchmark differently.
Li-Xuan
Qin, Richard P Beyer, Francesca N Hudson, Nancy J Linford, Daryl E Morris
and Kathleen F Kerr
Background: There are currently many different
methods for processing and summarizing probe level data from Affymetrix
oligonucleotide arrays. It is of great interest to validate these methods
and identify those that are most effective. There is no single best way
to do this validation, and a variety of approaches is needed. Moreover,
gene expression data are collected to answer a variety of scientific questions,
and the same method may not be best for all questions. Only a handful of
validation studies have been done so far, most of which rely on spike-in
datasets and focus on the question of detecting differential expression.
Here we seek methods that excel at estimating relative expression. We evaluate
methods by identifying those that give the strongest linear association
between expression measurements by array and the "gold-standard" assay.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
is generally considered the "gold-standard" assay for measuring
gene expression by biologists and is often used to confirm findings from
microarray data. Here we use qRT-PCR measurements to validate methods for
the components of processing oligo array data: background adjustment, normalization,
mismatch adjustment, and probeset summary. An advantage of our approach
over spike-in studies is that methods are validated on a real dataset that
was collected to address a scientific question.
Results: We initially identify three of six
popular methods that consistently produced the best agreement between oligo
array and RT-PCR data for medium- and high-intensity genes. The three methods
are generally known as MAS5, gcRMA, and the dChip mismatch mode. For medium-
and high-intensity genes, we identified use of data from mismatch probes
(as in MAS5 and dChip mismatch) and a sequence-based method of background
adjustment (as in gcRMA) as the most important factors in methods' performances.
However, we found poor reliability for methods using mismatch probes for
low-intensity genes, which is in agreement with previous studies. Conclusion:
We advocate use of sequence-based background adjustment in lieu of mismatch
adjustment to achieve the best results across the intensity spectrum. No
method of normalization or probeset summary showed any consistent advantages.
![]()
Peter
B Dallas, Nicholas G Gottardo, Martin J Firth, Alex H Beesley, Katrin Hoffmann,
Philippa A Terry, Joseph R Freitas, Joanne M Boag, Aaron J Cummings and
Ursula R Kees
Background: The use of microarray technology
to assess gene expression levels is now widespread in biology. The validation
of microarray results using independent mRNA quantitation techniques remains
a desirable element of any microarray experiment. To facilitate the comparison
of microarray expression data between laboratories it is essential that
validation methodologies be critically examined. We have assessed the correlation
between expression scores obtained for 48 human genes using oligonucleotide
microarrays and the expression levels for the same genes measured by quantitative
real-time RT-PCR (qRT-PCR).
Results: Correlations with qRT-PCR data were
obtained using microarray data that were processed using robust multi-array
analysis (RMA) and the MAS 5.0 algorithm. Our results indicate that when
identical transcripts are targeted by the two methods, correlations between
qRT-PCR and microarray data are generally strong (r = 0.89). However, we
observed poor correlations between qRT-PCR and RMA or MAS 5.0 normalized
microarray data for 13% or 16% of genes, respectively.
Conclusion: These results highlight the complementarity
of oligonucleotide microarray and qRTPCR technologies for validation of
gene expression measurements, while emphasizing the continuing requirement
for caution in interpreting gene expression data.
R. Sasik,
E. Calvo and J. Corbeil
High-density
oligonucleotide arrays (GeneChip, Affymetrix, Santa Clara, CA) have become
a standard research tool in many areas of biomedical research. They quantitatively
monitor the expression of thousands of genes simultaneously by measuring
fluorescence from gene-specific targets or probes. The relationship between
signal intensities and transcript abundance as well as normalization issues
have been the focus of much recent attention (Hill et al., 2001; Chudin
et al., 2002; Naef et al., 2002a). It is desirable that a researcher has
the best possible analytical tools to make the most of the information
that this powerful technology has to offer. At present there are three
analytical methods available: the newly released Affymetrix Microarray
Suite 5.0 (AMS) software that accompanies the GeneChip product, the method
of Li and Wong (LW; Li and Wong, 2001), and the method of Naef et al. (FN;
Naef et al., 2001). The AMS method is tailored for analysis of a single
microarray, and can therefore be used with any experimental design. The
LW method on the other hand depends on a large number of microarrays in
an experiment and cannot be used for an isolated microarray, and the FN
method is particular to paired microarrays, such as resulting from an experiment
in which each treatment sample has a corresponding control sample.
Our focus is on analysis of experiments in which there is a series of samples.
In this case only the AMS, LW, and the method described in this paper can
be used. The present method is model-based, like the LWmethod, but assumes
multiplicative not additive noise, and employs elimination of statistically
significant outliers for improved results. Unlike LW and AMS, we do not
assume probe-specific background (measured by the so-called mismatch probes).
Rather, we assume uniform background, whose level is estimated using both
the mismatch and perfect match probe intensities.
Kathe
E. Bjork and Karen Kafadar
Motivation: Affymetrix GeneChips are common
30 profiling platforms for quantifying gene expression. Using publicly
available datasets of expression profiles from human and mouse experiments,
we sought to characterize features of GeneChip data to better compare and
evaluate analyses for differential expression, regulation and clustering.
We uncovered an unexpected order dependence in expression data that holds
across a variety of chips in both human and mouse data.
Results: Order dependence among GeneChips
affected relative expression measures pre-processed and normalized with
the Affymetrix MAS5.0 algorithm and the robust multi-array average summarization
method. The effect strongly influenced detection calls and tests for differential
expression and can potentially significantly bias experimental results
based on GeneChip profiling.
![]()
Nandini
Raghavan, An M.I.M. De Bondt, Willem Talloen, Dieder Moechars, Hinrich
W.H. Göhlmann and Dhammika Amaratunga
Probe-level
data from Affymetrix GeneChips can be summarized in many ways to produce
probe-set level gene expression measures (GEMs). Disturbingly, the different
approaches not only generate quite different measures but they could also
yield very different analysis results. Here, we explore the question of
how much the analysis results really do differ, first at the gene level,
then at the biological process level. We demonstrate that, even though
the gene level results may not necessarily match each other particularly
well, as long as there is reasonably strong differentiation between the
groups in the data, the various GEMs do in fact produce results that are
similar to one another at the biological process level. Not only that the
results are biologically relevant. As the extent of differentiation drops,
the degree of concurrence weakens, although the biological relevance of
findings at the biological process level may yet remain.
Alexander
Statnikov, Constantin F. Aliferis, Ioannis Tsamardinos, Douglas Hardin
and Shawn Levy
Motivation: Cancer diagnosis is one of the most
important emerging clinical applications of gene expression microarray
technology.We are seeking to develop a computer system for powerful and
reliable cancer diagnostic model creation based on microarray data. To
keep a realistic perspective on clinical applications we focus on multicategory
diagnosis. To equip the system with the optimum combination of classifier,
gene selection and cross-validation methods, we performed a systematic
and comprehensive evaluation of several major algorithms for multicategory
classification, several gene selection methods, multiple ensemble classifier
methods and two cross-validation designs using 11 datasets spanning 74
diagnostic categories and 41 cancer types and 12 normal tissue types.
Results: Multicategory support vector machines
(MC-SVMs) are the most effective classifiers in performing accurate cancer
diagnosis from gene expression data. The MC-SVM techniques by Crammer and
Singer, Weston and Watkins and one-versus-rest were found to be the best
methods in this domain. MC-SVMs outperform other popular machine learning
algorithms, such as k-nearest neighbors, backpropagation and probabilistic
neural networks, often to a remarkable degree. Gene selection techniques
can significantly improve the classification performance of both MC-SVMs
and other non-SVM learning algorithms. Ensemble classifiers do not generally
improve performance of the best non-ensemble models. These results guided
the construction of a software system GEMS (Gene Expression Model Selector)
that automates high-quality model construction and enforces sound optimization
and performance estimation procedures. This is the first such system to
be informed by a rigorous comparative analysis of the available algorithms
and datasets.
Maïa Chanrion, Hélène Fontaine, Carmen Rodriguez, Vincent Negre, Frédéric Bibeau, Charles Theillet, Alain Hénaut and Jean-Marie Darbon
Background: Current histo-pathological prognostic
factors are not very helpful in predicting the clinical outcome of breast
cancer due to the disease's heterogeneity. Molecular profiling using a
large panel of genes could help to classify breast tumours and to define
signatures which are predictive of their clinical behaviour.
Methods: To this aim, quantitative RT-PCR
amplification was used to study the RNA expression levels of 47 genes in
199 primary breast tumours and 6 normal breast tissues. Genes were selected
on the basis of their potential implication in hormonal sensitivity of
breast tumours. Normalized RT-PCR data were analysed in an unsupervised
manner by pairwise hierarchical clustering, and the statistical relevance
of the defined subclasses was assessed by Chi2 analysis. The robustness
of the selected subgroups was evaluated by classifying an external and
independent set of tumours using these Chi2-defined molecular signatures.
Results: Hierarchical clustering of gene
expression data allowed us to define a series of tumour subgroups that
were either reminiscent of previously reported classifications, or represented
putative new subtypes. The Chi2 analysis of these subgroups allowed us
to define specific molecular signatures for some of them whose reliability
was further demonstrated by using the validation data set. A new breast
cancer subclass, called subgroup 7, that we defined in that way, was particularly
interesting as it gathered tumours with specific bioclinical features including
a low rate of recurrence during a 5 year follow-up.
Conclusion: The analysis of the expression of
47 genes in 199 primary breast tumours allowed classifying them into a
series of molecular subgroups. The subgroup 7, which has been highlighted
by our study, was remarkable as it gathered tumours with specific bioclinical
features including a low rate of recurrence. Although this finding should
be confirmed by using a larger tumour cohort, it suggests that gene expression
profiling using a minimal set of genes may allow the discovery of new subclasses
of breast cancer that are characterized by specific molecular signatures
and exhibit specific bioclinical features.
![]()
Frank
De Smet, Janick Mathys, Kathleen MarchaI, Gert Thijs, Bart De Moor and
Yves Moreau
Motivation: Microarray experiments generate
a considerable amount of data, which analyzed properly help us gain a huge
amount of biologically relevant information about the global cellular behaviour.
Clustering (grouping genes with similar expression profiles) is one of
the first steps in data analysis of high-throughput expression measurements.
A number of clustering algorithms have proved useful to make sense of such
data. These classical algorithms, though useful, suffer from several drawbacks
(e.g. they require the predefinition of arbitrary parameters like the number
of clusters; they force every gene into a cluster despite a low correlation
with other cluster members). ln the following we describe a novel adaptive
quality-based clustering algorithm that tackles some of these drawbacks.
Results: We propose a heuristic iterative
two-step algo- rithm: First, we find in the high-dimensional representation
of the data a sphere where the 'density' of expression profiles is locally
maximal (based on a preliminary estimate of the radius of the cluster-quality-based
approach). ln a second step, we derive an optimal radius of the cluster
(adaptive approach) so that only the significantly co-expressed genes are
included in the cluster. This estimation is achieved by fitting a model
to the data using an EM-algorithm. By inferring the radius from the data
itself, the biologist is freed from finding an optimal value for this radius
by trial-and-error. The computational complexity .of this method is approximately
linear in the number of gene expression profiles in the data set. Finally,
our method is successfully validated using existing data sets.
R. Keira
Curtis and Martin D. Brand
Motivation: Microarray experiments measure complex
changes in the abundance of many mRNAs under different conditions. Current
analysis methods cannot distinguish between direct and indirect effects
on expression, or calculate the relative importance of mRNAs in effecting
responses.
Results: Application of modular regulation
analysis to microarray data reveals and quantifies which mRNA changes are
important for cellular responses. The mRNAs are clustered, and then we
calculate how perturbations alter each cluster and how strongly those clusters
affect an output response. The product of these values quantifies how an
input changes a response through each cluster.
Two published
datasets are analysed. Two mRNA clusters transmit most of the response
of yeast doubling time to galactose; one contains mainly galactose metabolic
genes, and the other a regulatory gene. Analysis of the response of yeast
relative fitness to 2-deoxy-d-glucose reveals that control is distributed
between several mRNA clusters, but experimental error limits statistical
significance.
R. L.
Somorjai, B. Dolenko and R. Baumgartner
Motivation: Two practical realities constrain
the analysis of microarray data, mass spectra from proteomics, and biomedical
infrared or magnetic resonance spectra. One is the curse of dimensionality:
the number of features characterizing these data is in the thousands or
tens of thousands. The other is the curse of dataset sparsity: the number
of samples is limited. The consequences of these two curses are far-reaching
when such data are used to classify the presence or absence of disease.
Results: Using very simple classifiers, we
show for several publicly available microarray and proteomics datasets
how these curses influence classification outcomes. In particular, even
if the sample per feature ratio is increased to the recommended 510 by
feature extraction/reduction methods, dataset sparsity can render any classification
result statistically suspect. In addition, several optimal feature sets
are typically identifiable for sparse datasets, all producing perfect classification
results, both for the training and independent validation sets. This non-uniqueness
leads to interpretational difficulties and casts doubt on the biological
relevance of any of these optimal feature sets. We suggest an approach
to assess the relative quality of apparently equally good classifiers.
![]()
Michael B.
Eisen, Paul T. Spellman, Patrick O. Brown, and David Botstein
A system
of cluster analysis for genome-wide expression data from DNA microarray
hybridization is described that uses standard statistical algorithms to
arrange genes according to similarity in pattern of gene expression. The
output is displayed graphically, conveying the clustering and the underlying
expression data simultaneously in a form intuitive for biologists. We have
found in the budding yeast Saccharomyces cerevisiae that clustering gene
expression data groups together efficiently genes of known similar function,
and we find a similar tendency in human data. Thus patterns seen in genome-wide
expression experiments can be interpreted as indications of the status
of cellular processes. Also, coexpression of genes of known function with
poorly characterized or novel genes may provide a simple means of gaining
leads to the functions of many genes for which information is not available
currently.
Susmita
Datta and Somnath Datta
Motivation: With the advent of microarray chip
technology, large data sets are emerging containing the simultaneous expression
levels of thousands of genes at various time points during a biological
process. Biologists are attempting to group genes based on the temporal
pattern of their expression levels. While the use of hierarchical clustering
(UPGMA) with correlation distance has been the most common in the microarray
studies, there are many more choices of clustering algorithms in pattern
recognition and statistics literature. At the moment there do not seem
to be any clear-cut guidelines regarding the choice of a clustering algorithm
to be used for grouping genes based on their expression profiles.
Results: In this paper, we consider six clustering
algorithms (of various flavors!) and evaluate their performances on a well-known
publicly available microarray data set on sporulation of budding yeast
and on two simulated data sets. Among other things, we formulate three
reasonable validation strategies that can be used with any clustering algorithm
when temporal observations or replications are present. We evaluate each
of these six clustering methods with these validation measures. While the best method
is dependent on the exact validation strategy and the number of clusters
to be used, overall Diana appears to be a solid performer. Interestingly,
the performance of correlation-based hierarchical clustering and model-based
clustering (another method that has been advocated by a number of researchers)
appear to be on opposite extremes, depending on what validation measure
one employs. Next it is shown that the group means produced by Diana are
the closest and those produced by UPGMA are the farthest from a model profile
based on a set of hand-picked genes.
Daehee
Hwang, William A. Schmitt, George Stephanopoulos and Gregory Stephanopoulos
Motivation: Transcriptional profiling using
microarrays can reveal important information about cellular and tissue
expression phenotypes, but these measurements are costly and time consuming.
Additionally, tissue sample availability poses further constraints on the
number of arrays that can be analyzed in connection with a particular disease
or state of interest. It is therefore important to provide a method for
the determination of the minimum number of microarrays required to separate,
with statistical reliability, distinct disease states or other physiological
differences.
Results: Power analysis was applied to estimate
the minimum sample size required for two-class and multi-class discrimination.
The power analysis algorithm calculates the appropriate sample size for
discrimination of phenotypic subtypes in a reduced dimensional space obtained
by Fisher discriminant analysis (FDA). This approach was tested by applying
the algorithm to existing data sets for estimation of the minimum sample
size required for drawing certain conclusions on multi-class distinction
with statistical reliability. It was confirmed that when the minimum number
of samples estimated from power analysis is used, group means in the FDA
discrimination space are statistically different.
![]()
J. M.
Deutsch
Motivation: Microarray data has been shown recently
to be efficacious in distinguishing closely related cell types that often
appear in different forms of cancer, but is not yet practical clinically.
However, the data might be used to construct a minimal set of marker genes
that could then be used clinically by making antibody assays to diagnose
a specific type of cancer. Here a replication algorithm is used for this
purpose. It evolves an ensemble of predictors, all using different combinations
of genes to generate a set of optimal predictors.
Results: We apply this method to the leukemia
data of the Whitehead/MIT group that attempts to differentially diagnose
two kinds of leukemia, and also to data of Khan et al. to distinguish four
different kinds of childhood cancers. In the latter case we were able to
reduce the number of genes needed from 96 to less than 15, while at the
same time being able to classify all of their test data perfectly. We also
apply this method to two other cases, Diffuse large B-cell lymphoma data
(Shipp et al., 2002), and data of Ramaswamy et al. on multiclass diagnosis
of 14 common tumor types.
Ka Yee
Yeung, Mario Medvedovic and Roger E Bumgarner
Background: Cluster analysis is often used to
infer regulatory modules or biological function by associating unknown
genes with other genes that have similar expression patterns and known
regulatory elements or functions. However, clustering results may not have
any biological relevance.
Results: We applied various clustering algorithms
to microarray datasets with different sizes, and we evaluated the clustering
results by determining the fraction of gene pairs from the same clusters
that share at least one known common transcription factor. We used both
yeast transcription factor databases (SCPD, YPD) and chromatin immunoprecipitation
(ChIP) data to evaluate our clustering results. We showed that the ability
to identify co-regulated genes from clustering results is strongly dependent
on the number of microarray experiments used in cluster analysis and the
accuracy of these associations plateaus at between 50 and 100 experiments
on yeast data. Moreover, the model-based clustering algorithm MCLUST consistently
outperforms more traditional methods in accurately assigning co-regulated
genes to the same clusters on standardized data.
Conclusions: Our results are consistent with
respect to independent evaluation criteria that strengthen our confidence
in our results. However, when one compares ChIP data to YPD, the false-negative
rate is approximately 80% using the recommended p-value of 0.001. In addition,
we showed that even with large numbers of experiments, the false-positive
rate may exceed the truepositive rate. In particular, even when all experiments
are included, the best results produce clusters with only a 28% true-positive
rate using known gene transcription factor interactions.
Daniel
Berrar, Ian Bradbury and Werner Dubitzky
Background: Various statistical and machine
learning methods have been successfully applied to the classification of
DNA microarray data. Simple instance-based classifiers such as nearest
neighbor (NN) approaches perform remarkably well in comparison to more
complex models, and are currently experiencing a renaissance in the analysis
of data sets from biology and biotechnology. While binary classification
of microarray data has been extensively investigated, studies involving
multiclass data are rare. The question remains open whether there exists
a significant difference in performance between NN approaches and more
complex multiclass methods. Comparative studies in this field commonly
assess different models based on their classification accuracy only; however,
this approach lacks the rigor needed to draw reliable conclusions and is
inadequate for testing the null hypothesis of equal performance. Comparing
novel classification models to existing approaches requires focusing on
the significance of differences in performance.
Results: We investigated the performance
of instance-based classifiers, including a NN classifier able to assign
a degree of class membership to each sample. This model alleviates a major
problem of conventional instance-based learners, namely the lack of confidence
values for predictions. The model translates the distances to the nearest
neighbors into 'confidence scores'; the higher the confidence score, the
closer is the considered instance to a predefined class. We applied the
models to three real gene expression data sets and compared them with state-ofthe-
art methods for classifying microarray data of multiple classes, assessing
performance using a statistical significance test that took into account
the data resampling strategy. Simple NN classifiers performed as well as,
or significantly better than, their more intricate competitors.
Conclusion: Given its highly intuitive underlying
principles simplicity, ease-of-use, and robustness the k-NN classifier
complemented by a suitable distance-weighting regime constitutes an excellent
alternative to more complex models for multiclass microarray data sets.
Instance-based classifiers using weighted distances are not limited to
microarray data sets, but are likely to perform competitively in classifications
of high-dimensional biological data sets such as those generated by high-throughput
mass spectrometry.
![]()
Samuel
Kaski, Janne Nikkilä, Merja Oja, Jarkko Venna, Petri Törönen and Eero Castrén
Background: Conventionally, the first step in
analyzing the large and high-dimensional data sets measured by microarrays
is visual exploration. Dendrograms of hierarchical clustering, selforganizing
maps (SOMs), and multidimensional scaling have been used to visualize similarity
relationships of data samples. We address two central properties of the
methods: (i) Are the visualizations trustworthy, i.e., if two samples are
visualized to be similar, are they really similar? (ii) The metric. The
measure of similarity determines the result; we propose using a new learning
metrics principle to derive a metric from interrelationships among data
sets.
Results: The trustworthiness of hierarchical
clustering, multidimensional scaling, and the selforganizing map were compared
in visualizing similarity relationships among gene expression profiles.
The self-organizing map was the best except that hierarchical clustering
was the most trustworthy for the most similar profiles. Trustworthiness
can be further increased by treating separately those genes for which the
visualization is least trustworthy. We then proceed to improve the metric.
The distance measure between the expression profiles is adjusted to measure
differences relevant to functional classes of the genes. The genes for
which the new metric is the most different from the usual correlation metric
are listed and visualized with one of the visualization methods, the self-organizing
map, computed in the new metric.
Conclusions: The conjecture from the methodological
results is that the self-organizing map can be recommended to complement
the usual hierarchical clustering for visualizing and exploring gene expression
data. Discarding the least trustworthy samples and improving the metric
still improves it.
Laurent
Bréhélin, Olivier Gascuel and Olivier Martin
Motivation: Hierarchical clustering is a common
approach to study protein and gene expression data. This unsupervised technique
is used to find clusters of genes or proteins which are expressed in a
coordinated manner across a set of conditions. Because of both the biological
and technical variability, experimental repetitions are generally performed.
In this work, we propose an approach to evaluate the stability of clusters
derived from hierarchical clustering by taking repeated measurements into
account.
Results: The method is based on the bootstrap
technique that is used to obtain pseudo-hierarchies of genes from resampled
datasets. Based on a fast dynamic programming algorithm, we compare the
original hierarchy to the pseudo-hierarchies and assess the stability of
the original gene clusters. Then a shuffling procedure can be used to assess
the significance of the cluster stabilities. Our approach is illustrated
on simulated data and on two microarray datasets. Compared to the standard
hierarchical clustering methodology, it allows to point out the dubious
and stable clusters, and thus avoids misleading interpretations.
Vasyl
Pihur, Susmita Datta and Somnath Datta
Motivation: Biologists often employ clustering
techniques in the explorative phase of microarray data analysis to discover
relevant biological groupings. Given the availability of numerous clustering
algorithms in the machine-learning literature, an user might want to select
one that performs the best for his/her data set or application. While various
validation measures have been proposed over the years to judge the quality
of clusters produced by a given clustering algorithm including their biological
relevance, unfortunately, a given clustering algorithm can perform poorly
under one validation measure while outperforming many other algorithms
under another validation measure. A manual synthesis of results from multiple
validation measures is nearly impossible in practice, especially, when
a large number of clustering algorithms are to be compared using several
measures. An automated and objective way of reconciling the rankings is
needed.
Results: Using a Monte Carlo cross-entropy
algorithm, we successfully combine the ranks of a set of clustering algorithms
under consideration via a weighted aggregation that optimizes a distance
criterion. The proposed weighted rank aggregation allows for a far more
objective and automated assessment of clustering results than a simple
visual inspection. We illustrate our procedure using one simulated as well
as three real gene expression data sets from various platforms where we
rank a total of eleven clustering algorithms using a combined examination
of 10 different validation measures. The aggregate rankings were found
for a given number of clusters k and also for an entire range of k.
![]()
Yvan
Saeys, Iñaki Inza and Pedro Larrañaga
Feature
selection techniques have become an apparent need in many bioinformatics
applications. In addition to the large pool of techniques that have already
been developed in the machine learning and data mining fields, specific
applications in bioinformatics have led to a wealth of newly proposed techniques.
In this article, we make the interested reader aware of the possibilities
of feature selection, providing a basic taxonomy of feature selection techniques,
and discussing their use, variety and potential in a number of both common
as well as upcoming bioinformatics applications.
Zoltan
Kutalik, Jacqueline Inwald, Steve V. Gordon, R. Glyn Hewinson, Philip Butcher,
Jason Hinds, Kwang-Hyun Cho and Olaf Wolkenhauer
Motivation: When analyzing microarray data,
non-biological variation introduces uncertainty in the analysis and interpretation.
In this paper we focus on the validation of significant differences in
gene expression levels, or normalized channel intensity levels with respect
to different experimental conditions and with replicated measurements.
A myriad of methods have been proposed to study differences in gene expression
levels and to assign significance values as a measure of confidence. In
this paper we compare several methods, including SAM, regularized t -test,
mixture modeling, Wilks lambda score and variance stabilization. From
this comparison we developed a weighted resampling approach and applied
it to gene deletions in Mycobacterium bovis.
Results: We discuss the assumptions, model
structure, computational complexity and applicability to microarray data.
The results of our study justified the theoretical basis of the weighted
resampling approach, which clearly outperforms the others.
Martin
Vingron
The lab
biologist and theoretician need to make a concerted effort to design experiments
that can be realised and analysed. Bioinformaticians are predestined for
this role because they have learned to bridge the communication barriers
and they know the available data. But most of us need to improve the statistical
know-how or learn to efficiently interact with statisticians. The consequence
of all this is that we need to get back to school and learn more statistics.
Not so much with the goal of mastering all of statistics but with the goal
of sufficiently educating ourselves in order to pull in statisticians.
Alessandra Riva, Anne-Sophie Carpentier, Bruno Torrésani, Alain Hénaut
Microarrays
are becoming a ubiquitous tool of research in life sciences. However, theworking
principles of microarray-based methodologies are often misunderstood or
apparently ignored by the researchers who actually perform and interpret
experiments. This in turn seems to lead to a common over-expectation regarding
the explanatory and/or knowledge-generating power of microarray analyses.
In this
note we intend to explain basic principles of five (5) major groups of
analytical techniques used in studies of microarray data and their interpretation:
the principal component analysis (PCA), the independent component analysis
(ICA), the t-test, the analysis of variance (ANOVA), and self organizing
maps (SOM).We discuss answers to selected practical questions related to
the analysis of microarray data.We also take a closer look at the experimental
setup and the rules, which have to be observed in order to exploit microarrays
efficiently. Finally, we discuss in detail the scope and limitations of
microarray-based methods. We emphasize the fact that no amount of statistical
analysis can compensate for (or replace) a well thought through experimental
setup. We conclude that microarrays are indeed useful tools in life sciences
but by no means should they be expected to generate complete answers to
complex biological questions. We argue that even well posed questions,
formulated within a microarray-specific terminology, cannot be completely
answered with the use of microarray analyses alone.
![]()
Ian B
Jeffery, Desmond G Higgins and Aedín C Culhane
Background: Numerous feature selection methods
have been applied to the identification of differentially expressed genes
in microarray data. These include simple fold change, classical t-statistic
and moderated t-statistics. Even though these methods return gene lists
that are often dissimilar, few direct comparisons of these exist. We present
an empirical study in which we compare some of the most commonly used feature
selection methods. We apply these to 9 publicly available datasets, and
compare, both the gene lists produced and how these perform in class prediction
of test datasets.
Results: In this study, we compared the efficiency
of the feature selection methods; significance analysis of microarrays
(SAM), analysis of variance (ANOVA), empirical bayes t-statistic, template
matching, maxT, between group analysis (BGA), Area under the receiver operating
characteristic (ROC) curve, the Welch t-statistic, fold change, rank products,
and sets of randomly selected genes. In each case these methods were applied
to 9 different binary (two class) microarray datasets. Firstly we found
little agreement in gene lists produced by the different methods. Only
8 to 21% of genes were in common across all 10 feature selection methods.
Secondly, we evaluated the class prediction efficiency of each gene list
in training and test cross-validation using four supervised classifiers.
Conclusion: We report that the choice of feature
selection method, the number of genes in the gene list, the number of cases
(samples) and the noise in the dataset, substantially influence classification
success. Recommendations are made for choice of feature selection. Area
under a ROC curve performed well with datasets that had low levels of noise
and large sample size. Rank products performs well when datasets had low
numbers of samples or high levels of noise. The Empirical bayes t-statistic
performed well across a range of sample sizes.
Anbupalam
Thalamuthu, Indranil Mukhopadhyay, Xiaojing Zheng and George C. Tseng
Motivation: Microarray technology has been widely
applied in biological and clinical studies for simultaneous monitoring
of gene expression in thousands of genes. Gene clustering analysis is found
useful for discovering groups of correlated genes potentially co-regulated
or associated to the disease or conditions under investigation. Many clustering
methods including hierarchical clustering, K-means, PAM, SOM, mixture model-based
clustering and tight clustering have been widely used in the literature.
Yet no comprehensive comparative study has been performed to evaluate the
effectiveness of these methods.
Results: In this paper, six gene clustering
methods are evaluated by simulated data from a hierarchical log-normal
model with various degrees of perturbation as well as four real datasets.
A weighted Rand index is proposed for measuring similarity of two clustering
results with possible scattered genes (i.e. a set of noise genes not being
clustered). Performance of the methods in the real data is assessed by
a predictive accuracy analysis through verified gene annotations. Our results
show that tight clustering and model-based clustering consistently outperform
other clustering methods both in simulated and real data while hierarchical
clustering and SOM perform among the worst. Our analysis provides deep
insight to the complicated gene clustering problem of expression profile
and serves as a practical guideline for routine microarray cluster analysis.
Patrik
Rydén, Henrik Andersson, Mattias Landfors, Linda Näslund, Blanka Hartmanová,
Laila Noppa, and Anders Sjöstedt
Background: Recently, a large number of methods
for the analysis of microarray data have been proposed but there are few
comparisons of their relative performances. By using so-called spike-in
experiments, it is possible to characterize the analyzed data and thereby
enable comparisons of different analysis methods.
Results: A spike-in experiment using eight
in-house produced arrays was used to evaluate established and novel methods
for filtration, background adjustment, scanning, channel adjustment, and
censoring. The S-plus package EDMA, a stand-alone tool providing characterization
of analyzed cDNA-microarray data obtained from spike-in experiments, was
developed and used to evaluate 252 normalization methods. For all analyses,
the sensitivities at low false positive rates were observed together with
estimates of the overall bias and the standard deviation. In general, there
was a trade-off between the ability of the analyses to identify differentially
expressed genes (i.e. the analyses' sensitivities) and their ability to
provide unbiased estimators of the desired ratios. Virtually all analysis
underestimated the magnitude of the regulations; often less than 50% of
the true regulations were observed. Moreover, the bias depended on the
underlying mRNA-concentration; low concentration resulted in high bias.
Many of the analyses had relatively low sensitivities, but analyses that
used either the constrained model (i.e. a procedure that combines data
from several scans) or partial filtration (a novel method for treating
data from so-called not-found spots) had with few exceptions high sensitivities.
These methods gave considerable higher sensitivities than some commonly
used analysis methods.
Conclusion: The use of spike-in experiments
is a powerful approach for evaluating microarray preprocessing procedures.
Analyzed data are characterized by properties of the observed log-ratios
and the analysis' ability to detect differentially expressed genes. If
bias is not a major problem; we recommend the use of either the CM-procedure
or partial filtration.
![]()
Caroline
Truntzer, Catherine Mercier, Jacques Estève, Christian Gautier and Pascal
Roy
Background: With the advance of microarray technology,
several methods for gene classification and prognosis have been already
designed. However, under various denominations, some of these methods have
similar approaches. This study evaluates the influence of gene expression
variance structure on the performance of methods that describe the relationship
between gene expression levels and a given phenotype through projection
of data onto discriminant axes.
Results: We compared Between-Group Analysis
and Discriminant Analysis (with prior dimension reduction through Partial
Least Squares or Principal Components Analysis). A geometric approach showed
that these two methods are strongly related, but differ in the way they
handle data structure. Yet, data structure helps understanding the predictive
efficiency of these methods. Three main structure situations may be identified.
When the clusters of points are clearly split, both methods perform equally
well. When the clusters superpose, both methods fail to give interesting
predictions. In intermediate situations, the configuration of the clusters
of points has to be handled by the projection to improve prediction. For
this, we recommend Discriminant Analysis. Besides, an innovative way of
simulation generated the three main structures by modelling different partitions
of the whole variance into within-group and between-group variances. These
simulated datasets were used in complement to some well-known public datasets
to investigate the methods behaviour in a large diversity of structure
situations. To examine the structure of a dataset before analysis and preselect
an a priori appropriate method for its analysis, we proposed a two-graph
preliminary visualization tool: plotting patients on the Between- Group
Analysis discriminant axis (x-axis) and on the first and the second within-group
Principal Components Analysis component (y-axis), respectively.
Conclusion: Discriminant Analysis outperformed
Between-Group Analysis because it allows for the dataset structure. An
a priori knowledge of that structure may guide the choice of the analysis
method. Simulated datasets with known properties are valuable to assess
and compare the performance of analysis methods, then implementation on
real datasets checks and validates the results. Thus, we warn against the
use of unchallenging datasets for method comparison, such as the Golub
dataset, because their structure is such that any method would be efficient.
Wolfram
Weckwerth
The primary
aim of omic technologies is the nontargeted identification of all gene
products (transcripts, proteins, and metabolites) present in a specific
biological sample. By their nature, these technologies reveal unexpected
properties of biological systems. A second and more challenging aspect
of omic technologies is the refined analysis of quantitative dynamics in
biological systems. For metabolomics, gas and liquid chromatography coupled
to mass spectrometry are well suited for coping with high sample numbers
in reliable measurement times with respect to both technical accuracy and
the identification and quantitation of small-molecular-weight metabolites.
This potential is a prerequisite for the analysis of dynamic systems. Thus,
metabolomics is a key technology for systems biology.
The aim
of this review is to (a) provide an in-depth overview about metabolomic
technology, (b) explore how metabolomic networks can be connected to the
underlying reaction pathway structure, and (c) discuss the need to investigate
integrative biochemical networks.
David B. Allison, Xiangqin Cui, Grier P. Page and Mahyar Sabripour
In just
a few years, microarrays have gone from obscurity to being almost ubiquitous
in biological research. At the same time, the statistical methodology for
microarray analysis has progressed from simple visual assessments of results
to a weekly deluge of papers that describe purportedly novel algorithms
for analysing changes in gene expression. Although the many procedures
that are available might be bewildering to biologists who wish to apply
them, statistical geneticists are recognizing commonalities among the different
methods. Many are special cases of more general models, and points of consensus
are emerging about the general approaches that warrant use and elaboration.
Chodziwadziwa
Whiteson Kabudula
Progress
in microarray gene expression technology has been complemented by advances
in techniques and tools for microarray data analysis. There exist various
types of analyses of microarray data and a variety of public tools are
available for performing these analyses. Here, we present an overview of
three publicly-accessible web-based tools for microarray data analysis;
Gene Expression Pattern Analysis Suite (GEPAS), Expression Profiler: Next
Generation (EP:NG), and Microarray Data Analysis Web Tool (MIDAW). The
discussion particularly focuses on one of the most widely used microarray
data analysis techniques known as clustering. Insights are provided on
the properties and usefulness of each of the three tools with regard to
clustering. For each of the tools, a thorough exploration of the possibilities
provided for various clustering techniques is made. In addition, we present
a comparison analysis of the performance of the three tools with emphasis
on clustering.
![]()
Anne-Sophie Carpentier, Alessandra Riva, Pierre Tisseur, Gilles Didier, Alain Hénaut
The number
of statistical tools used to analyze transcriptome data is continuously
increasing and no one, definitive method has so far emerged. There is a
need for comparison and a number of different approaches has been taken
to evaluate the effectiveness of the different statistical tools available
for microarray analyses.
In this
paper, we describe a simple and efficient protocol to compare the reliability
of different statistical tools available for microarray analyses. It exploits
the fact that genes within an operon exhibit the same expression patterns.
In order to compare the tools, the genes are ranked according to the most
relevant criterion for each tool; for each tool we look at the number of
different operons represented within the first twenty genes detected. We
then look at the size of the interval within which we find the most significant
genes belonging to each operon in question. This allows us to define and
estimate the sensitivity and accuracy of each statistical tool.
We have
compared four statistical tools using Bacillus subtilis expression data:
the analysis of variance (ANOVA), the principal component analysis (PCA),
the independent component analysis (ICA) and the partial least square regression
(PLS). Our results show ICA to be the most sensitive and accurate of the
tools tested. In this article, we have used the protocol to compare statistical
tools applied to the analysis of differential gene expression. However,
it can also be applied without modification to compare the statistical
tools developed for other types of transcriptome analyses, like the study
of gene co-expression.
Guy N
Brock, John R Shaffer, Richard E Blakesley, Meredith J Lotz and George
C Tseng
Background: Gene expression data frequently
contain missing values, however, most downstream analyses for microarray
experiments require complete data. In the literature many methods have
been proposed to estimate missing values via information of the correlation
patterns within the gene expression matrix. Each method has its own advantages,
but the specific conditions for which each method is preferred remains
largely unclear. In this report we describe an extensive evaluation of
eight current imputation methods on multiple types of microarray experiments,
including time series, multiple exposures, and multiple exposures × time
series data. We then introduce two complementary selection schemes for
determining the most appropriate imputation method for any given data set.
Results: We found that the optimal imputation
algorithms (LSA, LLS, and BPCA) are all highly competitive with each other,
and that no method is uniformly superior in all the data sets we examined.
The success of each method can also depend on the underlying "complexity" of
the expression data, where we take complexity to indicate the difficulty
in mapping the gene expression matrix to a lower-dimensional subspace.
We developed an entropy measure to quantify the complexity of expression
matrixes and found that, by incorporating this information, the entropy
based selection (EBS) scheme is useful for selecting an appropriate imputation
algorithm. We further propose a simulation-based self-training selection
(STS) scheme. This technique has been used previously for microarray data
imputation, but for different purposes. The scheme selects the optimal
or near-optimal method with high accuracy but at an increased computational
cost.
Conclusion: Our findings provide insight into
the problem of which imputation method is optimal for a given data set.
Three top-performing methods (LSA, LLS and BPCA) are competitive with each
other. Global-based imputation methods (PLS, SVD, BPCA) performed better
on mcroarray data with lower complexity, while neighbour-based methods
(KNN, OLS, LSA, LLS) performed better in data with higher complexity. We
also found that the EBS and STS schemes serve as complementary and effective
tools for selecting the optimal imputation algorithm.
![]()
Mathieu
Miron, Owen Z Woody, Alexandre Marcil, Carl Murie, Robert Sladek and Robert
Nadon
Background: DNA microarrays are popular tools
for measuring gene expression of biological samples. This ever increasing
popularity is ensuring that a large number of microarray studies are conducted,
many of which with data publicly available for mining by other investigators.
Under most circumstances, validation of differential expression of genes
is performed on a gene to gene basis. Thus, it is not possible to generalize
validation results to the remaining majority of non-validated genes or
to evaluate the overall quality of these studies.
Results: We present an approach for the global
validation of DNA microarray experiments that will allow researchers to
evaluate the general quality of their experiment and to extrapolate validation
results of a subset of genes to the remaining non-validated genes. We illustrate
why the popular strategy of selecting only the most differentially expressed
genes for validation generally fails as a global validation strategy and
propose random-stratified sampling as a better gene selection method. We
also illustrate shortcomings of often-used validation indices such as overlap
of significant effects and the correlation coefficient and recommend the
concordance correlation coefficient (CCC) as an alternative.
Conclusion: We provide recommendations that
will enhance validity checks of microarray experiments while minimizing
the need to run a large number of labour-intensive individual validation
assays.
Huixia
Wang, Xuming He, Mark Band, Carole Wilson and Lei Liu
As gene
expression profile data from DNA microarrays accumulate rapidly, there
is a natural need to compare data across labs and platforms. Comparisons
of microarray data can be quite challenging due to data complexity and
variability. Different labs may adopt different technology platforms. One
may ask about the degree of agreement we can expect from different labs
and different platforms. To address this question, we conducted a study
of inter-lab and inter-platform agreement of microarray data across three
platforms and three labs. The statistical measures of consistency and agreement
used in this paper are the Pearson correlation, intraclass correlation,
kappa coefficients, and a measure of intra-transcript correlation. The
three platforms used in the present paper were Affymetrix GeneChip, custom
cDNA arrays, and custom oligo arrays. Using the within-platform variability
as a benchmark, we found that these technology platforms exhibited an acceptable
level of agreement, but the agreement between two technologies within the
same lab was greater than that between two labs using the same technology.
The consistency of replicates in each experiment varies from lab to lab.
When there is high consistency among replicates, different technologies
show good agreement within and across labs using the same RNA samples.
On the other hand, the lab effect, especially when confounded with the
RNA sample effect, plays a bigger role than the platform effect on data
agreement.
James
J. Chen Robert R. Delongchamp, Chen-An Tsai, Huey-miin Hsueh, Frank Sistare,
Karol L. Thompson, Varsha G. Desai and James C. Fuscoe
Motivation: A microarray experiment is a multi-step
process, and each step is a potential source of variation. There are two
major sources of variation: biological variation and technical variation.
This study presents a variance-components approach to investigating animal-to-animal,
between-array, within-array and day-to-day variations for two data sets.
The first data set involved estimation of technical variances for pooled
control and pooled treated RNA samples. The variance components included
between-array, and two nested within-array variances: between-section (the
upper- and lower sections of the array are replicates) and within-section
(two adjacent spots of the same gene are printed within each section).The
second experiment was conducted on four different weeks. Each week there
were reference and test samples with a dye-flip replicate in two hybridization
days. The variance components included week-to-week, animal-to-animal and
between-array and within-array variances.
Results: We applied the linear mixed-effects
model to quantify different sources of variation. In the first data set,
we found that the between-array variance is greater than the between-section
variance, which, in turn, is greater than the within-section variance.
In the second data set, for the reference samples, the week-to-week variance
is larger than the between-array variance, which, in turn, is slightly
larger than the within-array variance. For the test samples, the week-to-week
variance has the largest variation. The animal-to-animal variance is slightly
larger than the between-array and within-array variances. However, in a
gene-by-gene analysis, the animal-to-animal variance is smaller than the
between-array variance in four out of five housekeeping genes. In summary,
the largest variation observed is the week-to-week effect. Another important
source of variability is the animal-to-animal variation. Finally, we describe
the use of variance-component estimates to determine optimal numbers of
animals, arrays per animal and sections per array in planning microarray
experiments.
![]()
Kellie
J Archer, Catherine I Dumur, G Scott Taylor, Michael D Chaplin, Anthony
Guiseppi-Elie, Geraldine Grant, Andrea Ferreira-Gonzalez and Carleton T
Garrett
Background: Recent research examining cross-platform
correlation of gene expression intensities has yielded mixed results. In
this study, we demonstrate use of a correction factor for estimating cross-platform
correlations.
Results: In this paper, three technical replicate
microarrays were hybridized to each of three platforms. The three platforms
were then analyzed to assess both intra- and cross-platform reproducibility.
We present various methods for examining intra-platform reproducibility.
We also examine cross-platform reproducibility using Pearson's correlation.
Additionally, we previously developed a correction factor for Pearson's
correlation which is applicable when X and Y are measured with error. Herein
we demonstrate that correcting for measurement error by estimating the "disattenuated" correlation
substantially improves cross-platform correlations.
Conclusion: When estimating cross-platform correlation,
it is essential to thoroughly evaluate intra-platform reproducibility as
a first step. In addition, since measurement error is present in microarray
gene expression data, methods to correct for attenuation are useful in
decreasing the bias in cross-platform correlation estimates.
James
J Chen, Huey-Miin Hsueh, Robert R Delongchamp, Chien-Ju Lin and Chen-An
Tsai
Background: Many researchers are concerned with
the comparability and reliability of microarray gene expression data. Recent
completion of the MicroArray Quality Control (MAQC) project provides a
unique opportunity to assess reproducibility across multiple sites and
the comparability across multiple platforms. The MAQC analysis presented
for the conclusion of inter- and intra-platform comparability/reproducibility
of microarray gene expression measurements is inadequate. We evaluate the
reproducibility/comparability of the MAQC data for 12901 common genes in
four titration samples generated from five high-density one-color microarray
platforms and the TaqMan technology. We discuss some of the problems with
the use of correlation coefficient as metric to evaluate the inter- and
intraplatform reproducibility and the percent of overlapping genes (POG)
as a measure for evaluation of a gene selection procedure by MAQC.
Results: A total of 293 arrays were used
in the intra- and inter-platform analysis. A hierarchical cluster analysis
shows distinct differences in the measured intensities among the five platforms.
A number of genes show a small fold-change in one platform and a large
fold-change in another platform, even though the correlations between platforms
are high. An analysis of variance shows thirty percent of gene expressions
of the samples show inconsistent patterns across the five platforms. We
illustrated that POG does not reflect the accuracy of a selected gene list.
A non-overlapping gene can be truly differentially expressed with a stringent
cut, and an overlapping gene can be non-differentially expressed with non-stringent
cutoff. In addition, POG is an unusable selection criterion. POG can increase
or decrease irregularly as cutoff changes; there is no criterion to determine
a cutoff so that POG is optimized.
Conclusion: Using various statistical methods
we demonstrate that there are differences in the intensities measured by
different platforms and different sites within platform. Within each platform,
the patterns of expression are generally consistent, but there is site-by-site
variability. Evaluation of data analysis methods for use in regulatory
decision should take no treatment effect into consideration, when there
is no treatment effect, "a fold-change cutoff with a non-stringent
p-value cutoff" could result in 100% false positive error selection.
Andrew
J Holloway, Alicia Oshlack, Dileepa S Diyagama, David DL Bowtell and Gordon
K Smyth
Background: Concerns are often raised about
the accuracy of microarray technologies and the degree of cross-platform
agreement, but there are yet no methods which can unambiguously evaluate
precision and sensitivity for these technologies on a whole-array basis.
Results: A methodology is described for evaluating
the precision and sensitivity of whole-genome gene expression technologies
such as microarrays. The method consists of an easy-to-construct titration
series of RNA samples and an associated statistical analysis using non-linear
regression. The method evaluates the precision and responsiveness of each
microarray platform on a wholearray basis, i.e., using all the probes,
without the need to match probes across platforms. An experiment is conducted
to assess and compare four widely used microarray platforms. All four platforms
are shown to have satisfactory precision but the commercial platforms are
superior for resolving differential expression for genes at lower expression
levels. The effective precision of the two-color platforms is improved
by allowing for probe-specific dye-effects in the statistical model. The
methodology is used to compare three data extraction algorithms for the
Affymetrix platforms, demonstrating poor performance for the commonly used
proprietary algorithm relative to the other algorithms. For probes which
can be matched across platforms, the cross-platform variability is decomposed
into within-platform and between-platform components, showing that platform
disagreement is almost entirely systematic rather than due to measurement
variability.
Conclusion: The results demonstrate good precision
and sensitivity for all the platforms, but highlight the need for improved
probe annotation. They quantify the extent to which crossplatform measures
can be expected to be less accurate than within-platform comparisons for
predicting disease progression or outcome.
![]()
David
Petersen, GVR Chandramouli, Joel Geoghegan, Joanne Hilburn, Jonathon Paarlberg,
Chang Hee Kim, David Munroe, Lisa Gangi, Jing Han, Raj Puri, Lou Staudt,
John Weinstein, J Carl Barrett, Jeffrey Green and Ernest S Kawasaki
Background: Microarrays for the analysis of
gene expression are of three different types: short oligonucleotide (2530
base), long oligonucleotide (5080 base), and cDNA (highly variable in
length). The short oligonucleotide and cDNA arrays have been the mainstay
of expression analysis to date, but long oligonucleotide platforms are
gaining in popularity and will probably replace cDNA arrays. As part of
a validation study for the long oligonucleotide arrays, we compared and
contrasted expression profiles from the three formats, testing RNA from
six different cell lines against a universal reference standard.
Results: The three platforms had 6430 genes
in common. In general, correlation of gene expression levels across the
platforms was good when defined by concordance in the direction of expression
difference (upregulation or downregulation), scatter plot analysis, principal
component analysis, cell line correlation or quantitative RT-PCR. The overall
correlations (r values) between platforms were in the range 0.7 to 0.8,
as determined by analysis of scatter plots. When concordance was measured
for expression ratios significant at p-values of <0.05 and at expression
threshold levels of 1.5 and 2-fold, the agreement among the platforms was
very high, ranging from 93% to 100%.
Conclusion: Our results indicate that the long
oligonucleotide platform is highly suitable for expression analysis and
compares favorably with the cDNA and short oligonucleotide varieties. All
three platforms can give similar and reproducible results if the criterion
is the direction of change in gene expression and minimal emphasis is placed
on the magnitude of change.
Ann-Marie
Martoglio, James W. Miskin, Stephen K. Smith and David J.C. MacKay
Motivation: A number of algorithms and analytical
models have been employed to reduce the multidimensional complexity of
DNA array data and attempt to extract some meaningful interpretation of
the results. These include clustering, principal components analysis, self-organizing
maps, and support vector machine analysis. Each method assumes an implicit
model for the data, many of which separate genes into distinct clusters
defined by similar expression profiles in the samples tested. A point of
concern is that many genes may be involved in a number of distinct behaviours,
and should therefore be modelled to fit into as many separate clusters
as detected in the multidimensional gene expression space. The analysis
of gene expression data using a decomposition model that is independent
of the observer involved would be highly beneficial to improve standard
and reproducible classification of clinical and research samples.
Results: We present a variational independent
component analysis (ICA) method for reducing high dimensional DNA array
data to a smaller set of latent variables, each associated with a gene
signature. We present the results of applying the method to data from an
ovarian cancer study, revealing a number of tissue type-specific and tissue
type-independent gene signatures present in varying amounts among the samples
surveyed. The observer independent results of such molecular analysis of
biological samples could help identify patients who would benefit from
different treatment strategies. We further explore the application of the
model to similar highthroughput studies.
Lars
Gidskehaug, Endre Anderssen, Arnar Flatberg and Bjørn K Alsberg
Background: The most popular methods for significance
analysis on microarray data are well suited to find genes differentially
expressed across predefined categories. However, identification of features
that correlate with continuous dependent variables is more difficult using
these methods, and long lists of significant genes returned are not easily
probed for co-regulations and dependencies. Dimension reduction methods
are much used in the microarray literature for classification or for obtaining
low-dimensional representations of data sets. These methods have an additional
interpretation strength that is often not fully exploited when expression
data are analysed. In addition, significance analysis may be performed
directly on the model parameters to find genes that are important for any
number of categorical or continuous responses. We introduce a general scheme
for analysis of expression data that combines significance testing with
the interpretative advantages of the dimension reduction methods. This
approach is applicable both for explorative analysis and for classification
and regression problems.
Results: Three public data sets are analysed.
One is used for classification, one contains spiked-in transcripts of known
concentrations, and one represents a regression problem with several measured
responses. Model-based significance analysis is performed using a modified
version of Hotelling's T2-test, and a false discovery rate significance
level is estimated by resampling. Our results show that underlying biological
phenomena and unknown relationships in the data can be detected by a simple
visual interpretation of the model parameters. It is also found that measured
phenotypic responses may model the expression data more accurately than
if the design parameters are used as input. For the classification data,
our method finds much the same genes as the standard methods, in addition
to some extra which are shown to be biologically relevant. The list of
spiked-in genes is also reproduced with high accuracy.
Conclusion: The dimension reduction methods
are versatile tools that may also be used for significance testing. Visual
inspection of model components is useful for interpretation, and the methodology
is the same whether the goal is classification, prediction of responses,
feature selection or exploration of a data set. The presented framework
is conceptually and algorithmically simple, and a Matlab toolbox (Mathworks
Inc, USA) is supplemented.
![]()
Christelle Hennequet-Antier, Hélène Chiapello, Karine Piot, Séverine Degrelle, Isabelle Hue, Jean-Paul Renard, François Rodolphe and Stéphane Robin
Background: Analysis of variance is a powerful
approach to identify differentially expressed genes in a complex experimental
design for microarray and macroarray data. The advantage of the anova model
is the possibility to evaluate multiple sources of variation in an experiment.
Results: AnovArray is a package implementing
ANOVA for gene expression data using SAS® statistical software. The originality
of the package is 1) to quantify the different sources of variation on
all genes together, 2) to provide a quality control of the model, 3) to
propose two models for a gene's variance estimation and to perform a correction
for multiple comparisons.
Conclusion: AnovArray is freely available at http://www-mig.jouy.inra.fr/stat/AnovArray and
requires only SAS® statistical software.
Anat
Reiner-Benaim, Daniel Yekutieli, Noah E. Letwin, Gregory I. Elmer, Norman
H. Lee, Neri Kafkafi and Yoav Benjamini
Gene
expression and phenotypic functionality can best be associated when they
are measured quantitatively within the same experiment. The analysis of
such a complex experiment is presented, searching for associations between
measures of exploratory behavior in mice and gene expression in brain regions.
The analysis of such experiments raises several methodological problems.
First and foremost, the size of the pool of potential discoveries being
screened is enormous yet only few biologically relevant findings are expected,
making the problem of multiple testing especially severe. We present solutions
based on screening by testing related hypotheses, then testing the hypotheses
of interest. In one variant the subset is selected directly, in the other
one a tree of hypotheses is tested hierarchical; both variants control
the False Discovery Rate (FDR). Other problems in such experiments are
in the fact that the level of data aggregation may be different for the
quantitative traits (one per animal) and gene expression measurements (pooled
across animals); in that the association may not be linear; and in the
resolution of interest only few replications exist. We offer solutions
to these problems as well. The hierarchical FDR testing strategies presented
here can serve beyond the structure of our motivating example study to
any complex microarray study.
Susan
CP Renn, Nadia Aubin-Horth and Hans A Hofmann
Background:
Unravelling the path from genotype to phenotype, as it is influenced by
an organism's environment, is one of the central goals in biology. Gene
expression profiling by means of microarrays has become very prominent
in this endeavour, although resources exist only for relatively few model
systems. As genomics has matured into a comparative research program, expression
profiling now also provides a powerful tool for non-traditional model systems
to elucidate the molecular basis of complex traits.
Results:
Here we present a microarray constructed with ~4500 features, derived from
a brainspecific cDNA library for the African cichlid fish Astatotilapia
burtoni (Perciformes). Heterologous hybridization, targeting RNA to an
array constructed for a different species, is used for eight different
fish species. We quantified the concordance in gene expression profiles
across these species (number of genes and fold-changes). Although most
robust when target RNA is derived from closely related species (<10
MA divergence time), our results showed consistent profiles for other closely
related taxa (~65 MA divergence time) and, to a lesser extent, even very
distantly related species (>200 MA divergence time).
Conclusion:
This strategy overcomes some of the restrictions imposed on model systems
that are of importance for evolutionary and ecological studies, but for
which only limited sequence information is available. Our work validates
the use of expression profiling for functional genomics within a comparative
framework and provides a foundation for the molecular and cellular analysis
of complex traits in a wide range of organisms.
P. Chiappetta,
M.C. Roubaud and B. Torrésani
We develop
an approach for the exploratory analysis of gene expression data, based
upon blind source separation techniques. This approach exploits higher-order
statistics to identify a linear model for (logarithms of) expression profiles,
described as linear combinations of independent sources. As a result,
it yields elementary expression patterns (the sources), which may be
interpreted as potential regulation pathways. Further analysis of the so-obtained
sources show that they are generally characterized by a small number of
specific coexpressed or antiexpressed genes. In addition, the projections
of the expression profiles onto the estimated sources often provides significant
clustering of conditions. The algorithm relies on a large number of runs
of independent component analysis with random initializations, followed
by a search of consensus sources. It then provides estimates for independent
sources, together with an assessment of their robustness. The results obtained
on two datasets (namely, breast cancer data and Bacillus subtilis sulfur
metabolism data) show that some of the obtained gene families correspond
to well known families of coregulated genes, which validates the proposed
approach.
![]()
Kurt Fellenberg,
Nicole C. Hauser, Benedikt Brors, Albert Neutzner, Jörg D. Hoheisel, and
Martin Vingron
Correspondence
analysis is an explorative computational method for the study of associations
between variables. Much like principal component analysis, it displays
a low-dimensional projection of the data, e.g., into a plane. It does this,
though, for two variables simultaneously, thus revealing associations between
them. Here, we demonstrate the applicability of correspondence analysis
to and high value for the analysis of microarray data, displaying associations
between genes and experiments. To introduce the method,weshow its application
to the well-known Saccharomyces cerevisiae cell-cycle synchronization data
by Spellman et al. [Spellman, P. T., Sherlock, G., Zhang, M. Q., Iyer,
V. R., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher,
B. (1998) Mol. Biol. Cell 9, 32733297], allowing for comparison with their
visualization of this data set. Furthermore, we apply correspondence analysis
to a non-time-series data set of our own, thus supporting its general applicability
to microarray data of different complexity, underlying structure, and experimental
strategy (both two-channel fluorescence- tag and radioactive labeling).
Carsten
O Daub, Ralf Steuer, Joachim Selbig and Sebastian Kloska
Background: The information theoretic concept
of mutual information provides a general framework to evaluate dependencies
between variables. In the context of the clustering of genes with similar
patterns of expression it has been suggested as a general quantity of similarity
to extend commonly used linear measures. Since mutual information is defined
in terms of discrete variables, its application to continuous data requires
the use of binning procedures, which can lead to significant numerical
errors for datasets of small or moderate size.
Results: In this work, we propose a method
for the numerical estimation of mutual information from continuous data.
We investigate the characteristic properties arising from the application
of our algorithm and show that our approach outperforms commonly used algorithms:
The significance, as a measure of the power of distinction from random
correlation, is significantly increased. This concept is subsequently illustrated
on two large-scale gene expression datasets and the results are compared
to those obtained using other similarity measures. A C++ source code of
our algorithm is available for non-commercial use from kloska@scienion.de
upon request.
Conclusion: The utilisation of mutual information
as similarity measure enables the detection of non-linear correlations
in gene expression datasets. Frequently applied linear correlation measures,
which are often used on an ad-hoc basis without further justification,
are thereby extended.
Daniel
M. Kemp, N. R. Nirmala and Joseph D. Szustakowski
Motivation: We describe an extension of the
pathway-based enrichment approach for analyzing microarray data via a robust
test for transcriptional variance. The use of a variance test is intended
to identify additional patterns of transcriptional regulation in which
many genes in a pathway are up- and down-regulated. Such patterns may be
indicative of the reciprocal regulation of pathway activators and inhibitors
or of the differential regulation of separate biological sub-processes
and should extend the number of detectable patterns of transcriptional
modulation.
Results: We validated this new statistical
approach on a microarray experiment that captures the temporal transcriptional
profile of muscle differentiation in mouse C2C12 cells. Comparisons of
the transcriptional state of myoblasts and differentiated myotubes via
a robust variance test implicated several novel pathways in muscle cell
differentiation previously overlooked by a standard enrichment analysis.
Specifically, pathways involved in cell structure, calcium-mediated signaling
and muscle-specific signaling were identified as differentially modulated
based on their increased transcriptional variance. These biologically relevant
results validate this approach and demonstrate the flexible nature of pathway-based
methods of data analysis.
Agnieszka Sekowska, Stéphane Robin, Jean-Jacques Daudin, Alain Hénaut and Antoine Danchin
Background: In global gene expression profiling
experiments, variation in the expression of genes of interest can often
be hidden by general noise. To determine how biologically significant variation
can be distinguished under such conditions we have analyzed the differences
in gene expression when Bacillus subtilis is grown either on methionine
or on methylthioribose as sulfur source.
Results: An unexpected link between arginine
metabolism and sulfur metabolism was discovered, enabling us to identify
a high-affinity arginine transport system encoded by the yqiXYZ genes.
In addition, we tentatively identified a methionine/methionine sulfoxide
transport system which is encoded by the operon ytmIJKLMhisP and
is presumably used in the degradation of methionine sulfoxide to methane
sulfonate for sulfur recycling. Experimental parameters resulting in systematic
biases in gene expression were also uncovered. In particular, we found
that the late competence operons comE, comF and comG were
associated with subtle variations in growth conditions.
Conclusions: Using variance analysis it is possible
to distinguish between systematic biases and relevant gene-expression variation
in transcriptome experiments. Co-variation of metabolic gene expression
pathways was thus uncovered linking nitrogen and sulfur metabolism in B.
subtilis.
![]()
Gilles Didier, Pierre Brézellec, Elisabeth Rémy and Alain Hénaut
GeneANOVA
is an ANOVA-based software devoted to the analysis of gene expression data.
Rolf Turk,
Peter AC 't Hoen, Ellen Sterrenburg, Renée X de Menezes, Emile J de Meijer,
Judith M Boer, Gert-Jan B van Ommen and Johan T den Dunnen
Background: In this study, we investigated the
effect of genetic background on expression profiles. We analysed the transcriptome
of mouse hindlimb muscle of five frequently used mouse inbred strains using
spotted oligonucleotide microarrays.
Results: Through ANOVA analysis with a false
discovery rate of 10%, we show that 1.4% of the analysed genes is significantly
differentially expressed between these mouse strains. Differential expression
of several of these genes has been confirmed by quantitative RT-PCR. The
number of genes affected by genetic background is approximately ten-fold
lower than the number of differentially expressed genes caused by a dystrophic
genetic defect.
Conclusions: We conclude that evaluation of the
effect of background on gene expression profiles in the tissue under study
is an effective and sensible approach when comparing expression patterns
in animal models with heterogeneous genetic backgrounds. Genes affected
by the genetic background can be excluded in subsequent analyses of the
disease-related changes in expression profiles. This is often a more effective
strategy than backcrossing and inbreeding to obtain isogenic backgrounds.
Aapo
Hyvärinen and Erkki Oja
Independent
component analysis was originally developed to deal with problems that
are closely related to the cocktail party problem. Since the recent increase
of interest in ICA, it has become clear that this principle has a lot of
other interesting applications as well.
Attila
Frigyesi, Srinivas Veerla, David Lindgren and Mattias Höglund
Background: An alternative to standard approaches
to uncover biologically meaningful structures in micro array data is to
treat the data as a blind source separation (BSS) problem. BSS attempts
to separate a mixture of signals into their different sources and refers
to the problem of recovering signals from several observed linear mixtures.
In the context of micro array data, "sources" may correspond
to specific cellular responses or to co-regulated genes.
Results: We applied independent component
analysis (ICA) to three different microarray data sets; two tumor data
sets and one time series experiment. To obtain reliable components we used
iterated ICA to estimate component centrotypes. We found that many of the
low ranking components indeed may show a strong biological coherence and
hence be of biological significance. Generally ICA achieved a higher resolution
when compared with results based on correlated expression and a larger
number of gene clusters with significantly enriched for gene ontology (GO)
categories. In addition, components characteristic for molecular subtypes
and for tumors with specific chromosomal translocations were identified.
ICA also identified more than one gene clusters significant for the same
GO categories and hence disclosed a higher level of biological heterogeneity,
even within coherent groups of genes.
Conclusion: Although the ICA approach primarily
detects hidden variables, these surfaced as highly correlated genes in
time series data and in one instance in the tumor data. This further strengthens
the biological relevance of latent variables detected by ICA.
![]()
Wolfram
Liebermeister
Motivation: The expression of genes is controlled
by specific combinations of cellular variables. We applied Independent
Component Analysis (ICA) to gene expression data, deriving a linear model
based on hidden variables, which we term 'expression modes'. The expression
of each gene is a linear function of the expression modes, where, according
to the ICA model, the linear influences of different modes show a minimal
statistical dependence, and their distributions deviate sharply from the
normal distribution.
Results: Studying cell cycle-related gene
expression in yeast, we found that the dominant expression modes could
be related to distinct biological functions, such as phases of the cell
cycle or the mating response. Analysis of human lymphocytes revealed modes
that were related to characteristic differences between cell types. With
both data sets, the linear influences of the dominant modes showed distributions
with large tails, indicating the existence of specifically up- and downregulated
target genes. The expression modes and their influences can be used to
visualize the samples and genes in low dimensional spaces. A projection
to expression modes helps to highlight particular biological functions,
to reduce noise, and to compress the data in a biologically sensible way.
M. Scholz,
S. Gatzek, A. Sterling, O. Fiehn and J. Selbig
Motivation: Metabolite fingerprinting is a technology
for providing information from spectra of total compositions of metabolites.
Here, spectra acquisitions by microchip-based nanoflow-direct-infusion
QTOF mass spectrometry, a simple and high throughput technique, is tested
for its informative power. As a simple test case we are using Arabidopsis
thaliana crosses.The question is how metabolite fingerprinting reflects
the biological background. In many applications the classical principal
component analysis (PCA) is used for detecting relevant information. Here
a modern alternative is introducedthe independent component analysis (ICA).
Due to its independence condition, ICA is more suitable for our questions
than PCA.However, ICA has not been developed for a small number of high-dimensional
samples, therefore a strategy is needed to overcome this limitation.
Results:To apply ICA successfully it is essential
first to reduce the high dimension of the dataset, by using PCA. The number
of principal components determines the quality of ICA significantly, therefore
we propose a criterion for estimating the optimal dimension automatically.The
kurtosis measure is used to order the extracted components to our interest.
Applied to our A. thaliana data, ICA detects three relevant factors, two
biological and one technical, and clearly outperforms the PCA.
James
N Jarvis, Igor Dozmorov, Kaiyu Jiang, Mark Barton Frank, Peter Szodoray,
Philip Alex and Michael Centola
Juvenile
rheumatoid arthritis (JRA) has a complex, poorly characterized pathophysiology.
Modeling of transcriptosome behavior in pathologic specimens using microarrays
allows molecular dissection of complex autoimmune diseases. However, conventional
analyses rely on identifying statistically significant differences in gene
expression distributions between patients and controls. Since the principal
aspects of disease pathophysiology vary significantly among patients, these
analyses are biased. Genes with highly variable expression, those most
likely to regulate and affect pathologic processes, are excluded from selection,
as their distribution among healthy and affected individuals may overlap
significantly. Here we describe a novel method for analyzing microarray
data that assesses statistically significant changes in gene behavior at
the population level. This method was applied to expression profiles of
peripheral blood leukocytes from a group of children with polyarticular
JRA and healthy control subjects. Results from this method are compared
with those from a conventional analysis of differential gene expression
and shown to identify discrete subsets of functionally related genes relevant
to disease pathophysiology. These results reveal the complex action of
the innate and adaptive immune responses in patients and specifically underscore
the role of IFN-γ in disease pathophysiology. Discriminant function
analysis of data from a cohort of patients treated with conventional therapy
identified additional subsets of functionally related genes; the results
may predict treatment outcomes. While data from only 9 patients and 12
healthy controls was used, this preliminary investigation of the inflammatory
genomics of JRA illustrates the significant potential of utilizing complementary
sets of bioinformatics tools to maximize the clinical relevance of microarray
data from patients with autoimmune disease, even in small cohorts.
Virginia
Goss Tusher, Robert Tibshirani and Gilbert Chu
Microarrays
can measure the expression of thousands of genes to identify changes in
expression between different biological states. Methods are needed to determine
the significance of these changes while accounting for the enormous number
of genes. We describe a method, Significance Analysis of Microarrays (SAM),
that assigns a score to each gene on the basis of change in gene expression
relative to the standard deviation of repeated measurements. For genes
with scores greater than an adjustable threshold, SAM uses permutations
of the repeated measurements to estimate the percentage of genes identified
by chance, the false discovery rate (FDR). When the transcriptional response
of human cells to ionizing radiation was measured by microarrays, SAM identified
34 genes that changed at least 1.5-fold with an estimated FDR of 12%, compared
with FDRs of 60 and 84% by using conventional methods of analysis. Of the
34 genes, 19 were involved in cell cycle regulation and 3 in apoptosis.
Surprisingly, four nucleotide excision repair genes were induced, suggesting
that this repair pathway for UV-damaged DNA might play a previously unrecognized
role in repairing DNA damaged by ionizing radiation.
![]()
M. Kathleen
Kerr and Gary A. Churchill
Gene
expression microarrays are an innovative technology with enormous promise
to help geneticists explore and understand the genome. Although the potential
of this technology has been clearly demonstrated, many important and interesting
statistical questions persist. We relate certain features of microarrays
to other kinds of experimental data and argue that classical statistical
techniques are appropriate and useful. We advocate greater attention to
experimental design issues and a more prominent role for the ideas of statistical
inference in microarray studies.
Andrew
Whitehead and Douglas L Crawford
Background: Variation in gene expression is
extensive among tissues, individuals, strains, populations and species.
The interactions among these sources of variation are relevant for physiological
studies such as disease or toxic stress; for example, it is common for
pathologies such as cancer, heart failure and metabolic disease to be associated
with changes in tissue-specific gene expression or changes in metabolic
gene expression. But how conserved these differences are among outbred
individuals and among populations has not been well documented. To address
this we examined the expression of a selected suite of 192 metabolic genes
in brain, heart and liver in three populations of the teleost fish Fundulus
heteroclitus using a highly replicated experimental design.
Results: Half of the genes (48%) were differentially
expressed among individuals within a population-tissue group and 76% were
differentially expressed among tissues. Differences among tissues reflected
well established tissue-specific metabolic requirements, suggesting that
these measures of gene expression accurately reflect changes in proteins
and their phenotypic effects. Remarkably, only a small subset (31%) of
tissue-specific differences was consistent in all three populations.
Conclusions: These data indicate that many tissue-specific
differences in gene expression are unique to one population and thus are
unlikely to contribute to fundamental differences between tissue types.
We suggest that those subsets of treatment-specific gene expression patterns
that are conserved between taxa are most likely to be functionally related
to the physiological state in question.
Shunpu
Zhang
Background: The Significance Analysis of Microarrays
(SAM) is a popular method for detecting significantly expressed genes and
controlling the false discovery rate (FDR). Recently, it has been reported
in the literature that the FDR is not well controlled by SAM. Due to the
vast application of SAM in microarray data analysis, it is of great importance
to have an extensive evaluation of SAM and its associated R-package (sam2.20).
Results: Our study has identified several
discrepancies between SAM and sam2.20. One major difference is that SAM
and sam2.20 use different methods for estimating FDR. Such discrepancies
may cause confusion among the researchers who are using SAM or are developing
the SAM-like methods. We have also shown that SAM provides no meaningful
estimates of FDR and this problem has been corrected in sam2.20 by using
a different formula for estimating FDR. However, we have found that, even
with the improvement sam2.20 has made over SAM, sam2.20 may still produce
erroneous and even conflicting results under certain situations. Using
an example, we show that the problem of sam2.20 is caused by its use of
asymmetric cutoffs which are due to the large variability of null scores
at both ends of the order statistics. An obvious approach without the complication
of the order statistics is the conventional symmetric cutoff method. For
this reason, we have carried out extensive simulations to compare the performance
of sam2.20 and the symmetric cutoff method. Finally, a simple modification
is proposed to improve the FDR estimation of sam2.20 and the symmetric
cutoff method.
Conclusion: Our study shows that the most serious
drawback of SAM is its poor estimation of FDR. Although this drawback has
been corrected in sam2.20, the control of FDR by sam2.20 is still not satisfactory.
The comparison between sam2.20 and the symmetric cutoff method reveals
that the relative performance of sam2.20 to the symmetric cutff method
depends on the ratio of induced to repressed genes in a microarray data,
and is also affected by the ratio of DE to EE genes and the distributions
of induced and repressed genes. Numerical simulations show that the symmetric
cutoff method has the biggest advantage over sam2.20 when there are equal
number of induced and repressed genes (i.e., the ratio of induced to repressed
genes is 1). As the ratio of induced to repressed genes moves away from
1, the advantage of the symmetric cutoff method to sam2.20 is gradually
diminishing until eventually sam2.20 becomes significantly better than
the symmetric cutoff method when the differentially expressed (DE) genes
are either all induced or all repressed genes. Simulation results also
show that our proposed simple modification provides improved control of
FDR for both sam2.20 and the symmetric cutoff method.
![]()
Per Broberg
Background: In the analysis of microarray data
one generally produces a vector of p-values that for each gene give the
likelihood of obtaining equally strong evidence of change by pure chance.
The distribution of these p-values is a mixture of two components corresponding
to the changed genes and the unchanged ones. The focus of this article
is how to estimate the proportion unchanged and the false discovery rate
(FDR) and how to make inferences based on these concepts. Six published
methods for estimating the proportion unchanged genes are reviewed, two
alternatives are presented, and all are tested on both simulated and real
data. All estimates but one make do without any parametric assumptions
concerning the distributions of the p-values. Furthermore, the estimation
and use of the FDR and the closely related q-value is illustrated with
examples. Five published estimates of the FDR and one new are presented
and tested. Implementations in R code are available.
Results: A simulation model based on the
distribution of real microarray data plus two real data sets were used
to assess the methods. The proposed alternative methods for estimating
the proportion unchanged fared very well, and gave evidence of low bias
and very low variance. Different methods perform well depending upon whether
there are few or many regulated genes. Furthermore, the methods for estimating
FDR showed a varying performance, and were sometimes misleading. The new
method had a very low error.
Conclusion: The concept of the q-value or false
discovery rate is useful in practical research, despite some theoretical
and practical shortcomings. However, it seems possible to challenge the
performance of the published methods, and there is likely scope for further
developing the estimates of the FDR. The new methods provide the scientist
with more options to choose a suitable method for any particular experiment.
The article advocates the use of the conjoint information regarding false
positive and negative rates as well as the proportion unchanged when identifying
changed genes.
Roger
Higdon, Gerald van Belle and Eugene Kolker
The false
discovery rate (FDR) has been widely adopted to address the multiple comparisons
issue in high-throughput experiments such as microarray gene-expression
studies. However, while the FDR is quite useful as an approach to limit
false discoveries within a single experiment, like other multiple comparison
corrections it may be an inappropriate way to compare results across experiments.
This article uses several examples based on gene expression data to demonstrate
the potential misinterpretations that can arise from using FDR to compare
across experiments. Researchers should be aware of these pitfalls and wary
of using FDR to compare experimental results. FDR should be augmented with
other measures such as p-values and expression ratios. It is worth including
standard error and variance information for meta-analyses and, if possible,
the raw data for re-analyses. This is especially important for high-throughput
studies because data are often re-used for different objectives, including
comparing common elements across many experiments. No single error rate
or data summary may be appropriate for all of the different objectives.
Yang
Xie, Wei Pan and Arkady B. Khodursky
Motivation: False discovery rate (FDR) is defined
as the expected percentage of false positives among all the claimed positives.
In practice, with the true FDR unknown, an estimated FDR can serve as a
criterion to evaluate the performance of various statistical methods under
the condition that the estimated FDR approximates the true FDR well, or
at least, it does not improperly favor or disfavor any particular method.
Permutation methods have become popular to estimate FDR in genomic studies.
The purpose of this paper is 2-fold. First, we investigate theoretically
and empirically whether the standard permutation-based FDR estimator is
biased, and if so, whether the bias inappropriately favors or disfavors
any method. Second, we propose a simple modification of the standard permutation
to yield a better FDR estimator, which can in turn serve as a more fair
criterion to evaluate various statistical methods.
Results: Both simulated and real data examples
are used for illustration and comparison. Three commonly used test statistics,
the sample mean, SAM statistic and Students t-statistic, are considered.
The results show that the standard permutation method overestimates FDR.
The overestimation is the most severe for the sample mean statistic while
the least for the t-statistic with the SAM-statistic lying between the
two extremes, suggesting that one has to becautious when using the standard
permutation-based FDR estimates to evaluate various statistical methods.
In addition, our proposed FDR estimation method is simple and outperforms
the standard method.
![]()
Robert
Tibshirani
Background: In this short article, we discuss
a simple method for assessing sample size requirements in microarray experiments.
Results: Our method starts with the output
from a permutation-based analysis for a set of pilot data, e.g. from the
SAM package. Then for a given hypothesized mean difference and various
samples sizes, we estimate the false discovery rate and false negative
rate of a list of genes; these are also interpretable as per gene power
and type I error. We also discuss application of our method to other kinds
of response variables, for example survival outcomes.
Conclusion: Our method seems to be useful for
sample size assessment in microarray experiments.
Kyung
In Kim and Mark A van de Wiel
Background: We consider effects of dependence
among variables of high-dimensional data in multiple hypothesis testing
problems, in particular the False Discovery Rate (FDR) control procedures.
Recent simulation studies consider only simple correlation structures among
variables, which is hardly inspired by real data features. Our aim is to
systematically study effects of several network features like sparsity
and correlation strength by imposing dependence structures among variables
using random correlation matrices.
Results: We study the robustness against
dependence of several FDR procedures that are popular in microarray studies,
such as Benjamin-Hochberg FDR, Storey's q-value, SAM and resampling based
FDR procedures. False Non-discovery Rates and estimates of the number of
null hypotheses are computed from those methods and compared. Our simulation
study shows that methods such as SAM and the q-value do not adequately
control the FDR to the level claimed under dependence conditions. On the
other hand, the adaptive Benjamini-Hochberg procedure seems to be most
robust while remaining conservative. Finally, the estimates of the number
of true null hypotheses under various dependence conditions are variable.
Conclusion: We discuss a new method for efficient
guided simulation of dependent data, which satisfy imposed network constraints
as conditional independence structures. Our simulation setup allows for
a structural study of the effect of dependencies on multiple testing criterions
and is useful for testing a potentially new method on p0 or FDR estimation
in a dependency context.
Susmita
Datta and Somnath Datta
Motivation: Statistical tests for the detection
of differentially expressed genes lead to a large collection of p-values
one for each gene comparison. Without any further adjustment, these p-values
may lead to a large number of false positives, simply because the number
of genes to be tested is huge, which might mean wastage of laboratory resources.
To account for multiple hypotheses, these p-values are typically adjusted
using a single step method or a step-down method in order to achieve an
overall control of the error rate (the so-called familywise error rate).
In many applications, this may lead to an overly conservative strategy
leading to too few genes being flagged.
Results: In this paper we introduce a novel
empirical Bayes screening (EBS) technique to inspect a large number of
p-values in an effort to detect additional positive cases. In effect, each
case borrows strength from an overall picture of the alternative hypotheses
computed from all the p-values, while the entire procedure is calibrated
by a step-down method so that the familywise error rate at the complete
null hypothesis is still controlled. It is shown that the EBS has substantially
higher sensitivity than the standard step-down approach for multiple comparison
at the cost of a modest increase in the false discovery rate (FDR). The
EBS procedure also compares favorably when compared with existing FDR control
procedures formultiple testing. The EBS procedure is particularly useful
in situations where it is important to identify all possible potentially
positive cases which can be subjected to further confirmatory testing in
order to eliminate the false positives. We illustrated this screening procedure
using a data set on human colorectal cancer where we show that the EBS
method detected additional genes rela- ted to colon cancer that were missed
by other methods. This novel empirical Bayes procedure is advantageous
over our earlier proposed empirical Bayes adjustments due to the following
reasons: (i) it offers an automatic screening of the p-values the user
may obtain from a univariate (i.e., gene by gene) analysis package making
it extremely easy to use for a non-statistician, (ii) since it applies
to the p-values, the tests do not have to be t-tests; in particular they
could be F -tests which might arise in certain ANOVA formulations with
expression data or even nonparametric tests, (iii) the empirical Bayes
adjustment uses nonparametric function estimation techniques to estimate
the marginal density of the transformed p-values rather than using a parametric
model for the prior distribution and is therefore robust against model
mis-specification.
Hyuna
Yang and Gary Churchill
Motivation: Microarray data typically have small
numbers of observations per gene, which can result in low power for statistical
tests. Test statistics that borrow information from data across all of
the genes can improve power, but these statistics have non-standard distributions,
and their significance must be assessed using permutation analysis. When
sample sizes are small, the number of distinct permutations can be severely
limited, and pooling the permutation-derived test statistics across all
genes has been proposed. However, the null distribution of the test statistics
under permutation is not the same for equally and differentially expressed
genes. This can have a negative impact on both p-value estimation and the
power of information borrowing statistics.
Results: We investigate permutation based
methods for estimating pvalues. One of methods that uses pooling from a
selected subset of the data are shown to have the correct type I error
rate and to provide accurate estimates of the false discovery rate (FDR).
We provide guidelines to select an appropriate subset. We also demonstrate
that information borrowing statistics have substantially increased power
compared to the t-test in small experiments.
![]()
Peng
Liu and J. T. Gene Hwang
Motivation: Sample size calculation is important
in experimental design and is even more so in microarray or proteomic experiments
since only a few repetitions can be afforded. In the multiple testing problems
involving these experiments, it is more powerful and more reasonable to
control false discovery rate (FDR) or positive FDR (pFDR) instead of type
I error, e.g. family-wise error rate (FWER). When controlling FDR, the
traditional approach of estimating sample size by controlling type I error
is no longer applicable.
Results: Our proposed method applies to controlling
FDR. The sample size calculation is straightforward and requires minimal
computation, as illustrated with two sample t-tests and F-tests. Based
on simulation with the resultant sample size, the power is shown to be
achievable by the q-value procedure.
Franck Rapaport, Andrei Zinovyev, Marie Dutreix, Emmanuel Barillot and Jean-Philippe Vert
Background: Microarrays have become extremely
useful for analysing genetic phenomena, but establishing a relation between
microarray analysis results (typically a list of genes) and their biological
significance is often difficult. Currently, the standard approach is to
map a posteriori the results onto gene networks in order to elucidate the
functions perturbed at the level of pathways. However, integrating a priori
knowledge of the gene networks could help in the statistical analysis of
gene expression data and in their biological interpretation.
Results: We propose a method to integrate
a priori the knowledge of a gene network in the analysis of gene expression
data. The approach is based on the spectral decomposition of gene expression
profiles with respect to the eigenfunctions of the graph, resulting in
an attenuation of the high-frequency components of the expression profiles
with respect to the topology of the graph. We show how to derive unsupervised
and supervised classification algorithms of expression profiles, resulting
in classifiers with biological relevance. We illustrate the method with
the analysis of a set of expression profiles from irradiated and non-irradiated
yeast strains.
Conclusion: Including a priori knowledge of
a gene network for the analysis of gene expression data leads to good classification
performance and improved interpretability of the results.
Isabelle Rivals, Léon Personnaz, Lieng Taing and Marie-Claude Potier
Motivation: A number of available program packages
determine the significant enrichments and/or depletions of GO categories
among a class of genes of interest. Whereas a correct formulation of the
problem leads to a single exact null distribution, these GO tools use a
large variety of statistical tests whose denominations often do not clarify
the underlying P-value computations.
Summary: We review the different formulations
of the problem and the tests they lead to: the binomial, x2, equality of
two probabilities, Fishers exact and hypergeometric tests. We clarify
the relationships existing between these tests, in particular the equivalence
between the hypergeometric test and Fishers exact test. We recall that
the other tests are valid only for large samples, the test of equality
of two probabilities and the x2-test being equivalent. We discuss the appropriateness
of one- and two-sided P-values, as well as some discreteness and conservatism
issues.
![]()
Dan Nettleton,
Justin Recknor and James M. Reecy
Motivation: The field of microarray data analysis
is shifting emphasis from methods for identifying differentially expressed
genes to methods for identifying differentially expressed gene categories.
The latter approaches utilize a priori information about genes to group
genes into categories and enhance the interpretation of experiments aimed
at identifying expression differences across treatments. While almost all
of the existing approaches for identifying differentially expressed gene
categories are practically useful, they suffer from a variety of drawbacks.
Perhaps most notably, many popular tools are based exclusively on gene-specific
statistics that cannot detect many types of multivariate expression change.
Results: We have developed a nonparametric
multivariate method for identifying gene categories whose multivariate
expression distribution differs across two or more conditions. We illustrate
our approach and compare its performance to several existing procedures
via the analysis of a real data set and a unique data-based simulation
study designed to capture the challenges and complexities of practical
data analysis. We show that our method has good power for differentiating
between differentially expressed and nondifferentially expressed gene categories,
and we utilize a resampling based strategy for controling the false discovery
rate when testing multiple categories.
Purvesh
Khatri and Sorin Drăghici
Independent
of the platform and the analysis methods used, the result of a microarray
experiment is, in most cases, a list of differentially expressed genes.
An automatic ontological analysis approach has been recently proposed to
help with the biological interpretation of such results. Currently, this
approach is the de facto standard for the secondary analysis of high throughput
experiments and a large number of tools have been developed for this purpose.
We present a detailed comparison of 14 such tools using the following criteria:
scope of the analysis, visualization capabilities, statistical model(s)
used, correction for multiple comparisons, reference microarrays available,
installation issues and sources of annotation data. This detailed analysis
of the capabilities of these tools will help researchers choose the most
appropriate tool for a given type of analysis. More importantly, in spite
of the fact that this type of analysis has been generally adopted, this
approach has several important intrinsic drawbacks. These drawbacks are
associated with all tools discussed and represent conceptual limitations
of the current state-of-the-art in ontological analysis. We propose these
as challenges for the next generation of secondary data analysis tools.
![]()
Juliane Schäfer
and Korbinian Strimmer
Motivation: Genetic networks are often described
statistically using graphical models (e.g. Bayesian networks). However,
inferring the network structure offers a serious challenge in microarray
analysis where the sample size is small compared to the number of considered
genes. This renders many standard algorithms for graphical models inapplicable,
and inferring genetic networks an ill-posed inverse problem.
Methods: We introduce a novel framework for
small-sample inference of graphical models from gene expression data. Specifically,
we focus on the so-called graphical Gaussian models (GGMs) that are now
frequently used to describe gene association networks and to detect conditionally
dependent genes. Our new approach is based on (1) improved (regularized)
small-sample point estimates of partial correlation, (2) an exact test
of edge inclusion with adaptive estimation of the degree of freedom and
(3) a heuristic network search based on false discovery rate multiple testing.
Steps (2) and (3) correspond to an empirical Bayes estimate of the network
topology.
Results: Using computer simulations, we investigate
the sensitivity (power) and specificity (true negative rate) of the proposed
framework to estimate GGMs from microarray data. This shows that it is
possible to recover the true network topology with high accuracy even for
small-sample datasets. Subsequently, we analyze gene expression data from
a breast cancer tumor study and illustrate our approach by inferring a
corresponding large-scale gene association network for 3883 genes.
Jelle
J. Goeman and Peter Bühlmann
Motivation: Many statistical tests have been
proposed in recent years for analyzing gene expression data in terms of
gene sets, usually from Gene Ontology. These methods are based on widely
different methodological assumptions. Some approaches test differential
expression of each gene set against differential expression of the rest
of the genes, whereas others test each gene set on its own. Also, some
methods are based on a model in which the genes are the sampling units,
whereas others treat the subjects as the sampling units. This article aims
to clarify the assumptions behind different approaches and to indicate
a preferential methodology of gene set testing.
Results: We identify some crucial assumptions
which are needed by the majority of methods. P-values derived from methods
that use a model which takes the genes as the sampling unit are easily
misinterpreted, as they are based on a statistical model that does not
resemble the biological experiment actually performed. Furthermore, because
these models are based on a crucial and unrealistic independence assumption
between genes, the P-values derived from such methods can be wildly anti-conservative,
as a simulation experiment shows. We also argue that methods that competitively
test each gene set against the rest of the genes create an unnecessary
rift between single gene testing and gene set testing.
Qi Liu,
Irina Dinu, Adeniyi J Adewale, John D Potter and Yutaka Yasui
Background: Multiple data-analytic methods have
been proposed for evaluating gene-expression levels in specific biological
pathways, assessing differential expression associated with a binary phenotype.
Following Goeman and Bühlmann's recent review, we compared statistical
performance of three methods, namely Global Test, ANCOVA Global Test, and
SAM-GS, that test "self-contained null hypotheses" Via. subject
sampling. The three methods were compared based on a simulation experiment
and analyses of three real-world microarray datasets.
Results: In the simulation experiment, we
found that the use of the asymptotic distribution in the two Global Tests
leads to a statistical test with an incorrect size. Specifically, p-values
calculated by the scaled χ2 distribution of Global Test
and the asymptotic distribution of ANCOVA Global Test are too liberal,
while the asymptotic distribution with a quadratic form of the Global Test
results in p-values that are too conservative. The two Global Tests with
permutation-based inference, however, gave a correct size. While the three
methods showed similar power using permutation inference after a proper
standardization of gene expression data, SAM-GS showed slightly higher
power than the Global Tests. In the analysis of a real-world microarray
dataset, the two Global Tests gave markedly different results, compared
to SAM-GS, in identifying pathways whose gene expressions are associated
with p53 mutation in cancer cell lines. A proper standardization of gene
expression variances is necessary for the two Global Tests in order to
produce biologically sensible results. After the standardization, the three
methods gave very similar biologically-sensible results, with slightly
higher statistical significance given by SAM-GS. The three methods gave
similar patterns of results in the analysis of the other two microarray
datasets.
Conclusion: An appropriate standardization makes
the performance of all three methods similar, given the use of permutation-based
inference. SAM-GS tends to have slightly higher power in the lower a-level
region (i.e. gene sets that are of the greatest interest). Global Test
and ANCOVA Global Test have the important advantage of being able to analyze
continuous and survival phenotypes and to adjust for covariates. A free
Microsoft Excel Add-In to perform SAM-GS is available from http://www.ualberta.ca/~yyasui/homepage.html.
![]()
Masami
Yokota Hirai, Mitsuru Yano, Dayan B. Goodenowe, Shigehiko Kanaya, Tomoko
Kimura, Motoko Awazuhara, Masanori Arita, Toru Fujiwara, and Kazuki Saito
Plant
metabolism is a complex set of processes that produce a wide diversity
of foods, woods, and medicines. With the genome sequences of Arabidopsis
and rice in hands, postgenomics studies integrating all omics sciences
can depict precise pictures of a whole-cellular process. Here, we present,
to our knowledge, the first report of investigation for gene-to-metabolite
networks regulating sulfur and nitrogen nutrition and secondary metabolism
in Arabidopsis, with integration of metabolomics and transcriptomics. Transcriptome
and metabolome analyses were carried out, respectively, with DNA macroarray
and several chemical analytical methods, including ultra high-resolution
Fourier transform-ion cyclotron MS. Mathematical analyses, including principal
component analysis and batch-learning self-organizing map analysis of transcriptome
and metabolome data suggested the presence of general responses to sulfur
and nitrogen deficiencies. In addition, specific responses to either sulfur
or nitrogen deficiency were observed in several metabolic pathways: in
particular, the genes and metabolites involved in glucosinolate metabolism
were shown to be coordinately modulated. Understanding such geneto- metabolite
networks in primary and secondary metabolism through integration of transcriptomics
and metabolomics can lead to identification of gene function and subsequent
improvement of production of useful compounds in plants.
John
Tomfohr, Jun Lu and Thomas B Kepler
Background: A promising direction in the analysis
of gene expression focuses on the changes in expression of specific predefined
sets of genes that are known in advance to be related (e.g., genes coding
for proteins involved in cellular pathways or complexes). Such an analysis
can reveal features that are not easily visible from the variations in
the individual genes and can lead to a picture of expression that is more
biologically transparent and accessible to interpretation. In this article,
we present a new method of this kind that operates by quantifying the level
of 'activity' of each pathway in different samples. The activity levels,
which are derived from singular value decompositions, form the basis for
statistical comparisons and other applications.
Results: We demonstrate our approach using
expression data from a study of type 2 diabetes and another of the influence
of cigarette smoke on gene expression in airway epithelia. A number of
interesting pathways are identified in comparisons between smokers and
non-smokers including ones related to nicotine metabolism, mucus production,
and glutathione metabolism. A comparison with results from the related
approach, 'gene-set enrichment analysis', is also provided.
Conclusion: Our method offers a flexible basis
for identifying differentially expressed pathways from gene expression
data. The results of a pathway-based analysis can be complementary to those
obtained from one more focused on individual genes. A web program PLAGE
(Pathway Level Analysis of Gene Expression) for performing the kinds of
analyses described here is accessible at http://dulci.biostat.duke.edu/pathways.
![]()
Fangxin
Hong and Rainer Breitling
Motivation: The proliferation of public data
repositories creates a need for meta-analysis methods to efficiently evaluate,
integrate and validate related datasets produced by independent groups.
A t-based approach has been proposed to integrate effect size from multiple
studies by modeling both intra- and between-study variation. Recently,
a non-parametric rank product method, which is derived based on biological
reasoning of fold-change criteria, has been applied to directly combine
multiple datasets into one meta study. Fishers Inverse χ2 method,
which only depends on P-values from individual analyses of each dataset,
has been used in a couple of medical studies. While these methods address
the question from different angles, it is not clear how they compare with
each other.
Results: We comparatively evaluate the three
methods; t-based hierarchical modeling, rank products and Fishers Inverse χ2 test
with P-values from either the t-based or the rank product method. A simulation
study shows that the rank product method, in general, has higher sensitivity
and selectivity than the t-based method in both individual and meta-analysis,
especially in the setting of small sample size and/or large between-study
variation. Not surprisingly, Fishers χ2 method highly
depends on the method used in the individual analysis. Application to real
datasets demonstrates that meta-analysis achieves more reliable identification
than an individual analysis, and rank products are more robust in gene
ranking, which leads to a much higher reproducibility among independent
studies. Though t-based meta-analysis greatly improves over the individual
analysis, it suffers from a potentially large amount of false positives
when P-values serve as threshold. We conclude that careful meta-analysis
is a powerful tool for integrating multiple array studies.
Erin
M Conlon, Joon J Song and Anna Liu
Background: With the growing abundance of microarray
data, statistical methods are increasingly needed to integrate results
across studies. Two common approaches for meta-analysis of microarrays
include either combining gene expression measures across studies or combining
summaries such as p-values, probabilities or ranks. Here, we compare two
Bayesian meta-analysis models that are analogous to these methods.
Results: Two Bayesian meta-analysis models
for microarray data have recently been introduced. The first model combines
standardized gene expression measures across studies into an overall mean,
accounting for inter-study variability, while the second combines probabilities
of differential expression without combining expression values. Both models
produce the gene-specific posterior probability of differential expression,
which is the basis for inference. Since the standardized expression integration
model includes inter-study variability, it may improve accuracy of results
versus the probability integration model. However, due to the small number
of studies typical in microarray meta-analyses, the variability between
studies is challenging to estimate. The probability integration model eliminates
the need to model variability between studies, and thus its implementation
is more straightforward. We found in simulations of two and five studies
that combining probabilities outperformed combining standardized gene expression
measures for three comparison values: the percent of true discovered genes
in meta-analysis versus individual studies; the percent of true genes omitted
in meta-analysis versus separate studies, and the number of true discovered
genes for fixed levels of Bayesian false discovery. We identified similar
results when pooling two independent studies of Bacillus subtilis. We assumed
that each study was produced from the same microarray platform with only
two conditions: a treatment and control, and that the data sets were pre-scaled.
Conclusion: The Bayesian meta-analysis model
that combines probabilities across studies does not aggregate gene expression
measures, thus an inter-study variability parameter is not included in
the model. This results in a simpler modeling approach than aggregating
expression measures, which accounts for variability across studies. The
probability integration model identified more true discovered genes and
fewer true omitted genes than combining expression measures, for our data
sets.
Paola
Pedotti, Peter A.C. 't Hoen, Erno Vreugdenhil, Geert J. Schenk, Rolf H.A.M.
Vossen, Yavuz Ariyurek, Mattias de Hollander, Rowan Kuiper, Gertjan J.B.
van Ommen, Johan T. den Dunnen, Judith M. Boer, Renée X. de Menezes
Background: The comparability of gene expression
data generated with different microarray platforms is still a matter of
concern. Here we address the performance and the overlap in the detection
of differentially expressed genes for five different microarray platforms
in a challenging biological context where differences in gene expression
are few and subtle.
Results: Gene expression profiles in the
hippocampus of five wild-type and five transgenic δC-doublecortin-like
kinase mice were evaluated with five microarray platforms: Applied Biosystems,
Affymetrix, Agilent, Illumina, LGTC home-spotted arrays. Using a fixed
false discovery rate of 10% we detected surprising differences between
the number of differentially expressed genes per platform. Four genes were
selected by ABI, 130 by Affymetrix, 3,051 by Agilent, 54 by Illumina, and
13 by LGTC. Two genes were found significantly differentially expressed
by all platforms and the four genes identified by the ABI platform were
found by at least three other platforms. Quantitative RT-PCR analysis confirmed
20 out of 28 of the genes detected by two or more platforms and 8 out of
15 of the genes detected by Agilent only. We observed improved correlations
between platforms when ranking the genes based on the significance level
than with a fixed statistical cut-off. We demonstrate significant overlap
in the affected gene sets identified by the different platforms, although
biological processes were represented by only partially overlapping sets
of genes. Aberrances in GABA-ergic signalling in the transgenic mice were
consistently found by all platforms.
Conclusions: The different microarray platforms
give partially complementary views on biological processes affected. Our
data indicate that when analyzing samples with only subtle differences
in gene expression the use of two different platforms might be more attractive
than increasing the number of replicates. Commercial two-color platforms
seem to have higher power for finding differentially expressed genes between
groups with small differences in expression.
![]()
Homin
K. Lee, Amy K. Hsu, Jon Sajdak, Jie Qin, and Paul Pavlidis
We present
a large-scale analysis of mRNA coexpression based on 60 large human data
sets containing a total of 3924 microarrays. We sought pairs of genes that
were reliably coexpressed (based on the correlation of their expression
profiles) in multiple data sets, establishing a high-confidence network
of 8805 genes connected by 220,649 coexpression links that are observed
in at least three data sets. Confirmed positive correlations between genes
were much more common than confirmed negative correlations. We show that
confirmation of coexpression in multiple data sets is correlated with functional
relatedness, and show how cluster analysis of the network can reveal functionally
coherent groups of genes. Our findings demonstrate how the large body of
accumulated microarray data can be exploited to increase the reliability
of inferences about gene function.
John
R Stevens and RW Doerge
Background: As the use of microarray technology
becomes more prevalent it is not unusual to find several laboratories employing
the same microarray technology to identify genes related to the same condition
in the same species. Although the experimental specifics are similar, typically
a different list of statistically significant genes result from each data
analysis.
Results: We propose a statistically-based
meta-analytic approach to microarray analysis for the purpose of systematically
combining results from the different laboratories. This approach provides
a more precise view of genes that are significantly related to the condition
of interest while simultaneously allowing for differences between laboratories.
Of particular interest is the widely used Affymetrix oligonucleotide array,
the results of which are naturally suited to a meta-analysis. A simulation
model based on the Affymetrix platform is developed to examine the adaptive
nature of the meta-analytic approach and to illustrate the usefulness of
such an approach in combining microarray results across laboratories. The
approach is then applied to real data involving a mouse model for multiple
sclerosis.
Conclusion: The quantitative estimates from
the meta-analysis model tend to be closer to the "true" degree
of differential expression than any single lab. Meta-analytic methods can
systematically combine Affymetrix results from different laboratories to
gain a clearer understanding of genes' relationships to specific conditions
of interest.
Hongying
Jiang, Youping Deng, Huann-Sheng Chen, Lin Tao, Qiuying Sha, Jun Chen,
Chung-Jui Tsai and Shuanglin Zhang
Background: Due to the high cost and low reproducibility
of many microarray experiments, it is not surprising to find a limited
number of patient samples in each study, and very few common identified
marker genes among different studies involving patients with the same disease.
Therefore, it is of great interest and challenge to merge data sets from
multiple studies to increase the sample size, which may in turn increase
the power of statistical inferences. In this study, we combined two lung
cancer studies using micorarray GeneChip®, employed two gene shaving methods
and a two-step survival test to identify genes with expression patterns
that can distinguish diseased from normal samples, and to indicate patient
survival, respectively.
Results: In addition to common data transformation
and normalization procedures, we applied a distribution transformation
method to integrate the two data sets. Gene shaving (GS) methods based
on Random Forests (RF) and Fisher's Linear Discrimination (FLD) were then
applied separately to the joint data set for cancer gene selection. The
two methods discovered 13 and 10 marker genes (5 in common), respectively,
with expression patterns differentiating diseased from normal samples.
Among these marker genes, 8 and 7 were found to be cancer-related in other
published reports. Furthermore, based on these marker genes, the classifiers
we built from one data set predicted the other data set with more than
98% accuracy. Using the univariate Cox proportional hazard regression model,
the expression patterns of 36 genes were found to be significantly correlated
with patient survival (p < 0.05). Twenty-six of these 36 genes were
reported as survival-related genes from the literature, including 7 known
tumor-suppressor genes and 9 oncogenes. Additional principal component
regression analysis further reduced the gene list from 36 to 16.
Conclusion: This study provided a valuable method
of integrating microarray data sets with different origins, and new methods
of selecting a minimum number of marker genes to aid in cancer diagnosis.
After careful data integration, the classification method developed from
one data set can be applied to the other with high prediction accuracy.
![]()
Andrey
A. Shabalin, Håkon Tjelmeland, Cheng Fan, Charles M. Perou and Andrew B.
Nobel
Motivation: Gene-expression microarrays are
currently being applied in a variety of biomedical applications. This article
considers the problem of how to merge datasets arising from different gene-expression
studies of a common organism and phenotype. Of particular interest is how
to merge data from different technological platforms.
Results: The article makes two contributions
to the problem. The first is a simple cross-study normalization method,
which is based on linked gene/sample clustering of the given datasets.
The second is the introduction and description of several general validation
measures that can be used to assess and compare cross-study normalization
methods. The proposed normalization method is applied to three existing
breast cancer datasets, and is compared to several competing normalization
methods using the proposed validation measures.
Andrew
Whitehead and Douglas L Crawford
Background: Variation in gene expression is
extensive among tissues, individuals, strains, populations and species.
The interactions among these sources of variation are relevant for physiological
studies such as disease or toxic stress; for example, it is common for
pathologies such as cancer, heart failure and metabolic disease to be associated
with changes in tissue-specific gene expression or changes in metabolic
gene expression. But how conserved these differences are among outbred
individuals and among populations has not been well documented. To address
this we examined the expression of a selected suite of 192 metabolic genes
in brain, heart and liver in three populations of the teleost fish Fundulus
heteroclitus using a highly replicated experimental design.
Results: Half of the genes (48%) were differentially
expressed among individuals within a population-tissue group and 76% were
differentially expressed among tissues. Differences among tissues reflected
well established tissue-specific metabolic requirements, suggesting that
these measures of gene expression accurately reflect changes in proteins
and their phenotypic effects. Remarkably, only a small subset (31%) of
tissue-specific differences was consistent in all three populations.
Conclusions: These data indicate that many tissue-specific
differences in gene expression are unique to one population and thus are
unlikely to contribute to fundamental differences between tissue types.
We suggest that those subsets of treatment-specific gene expression patterns
that are conserved between taxa are most likely to be functionally related
to the physiological state in question.
![]()
Xin Gao
and Peter XK Song
Background: Numerous nonparametric approaches
have been proposed in literature to detect differential gene expression
in the setting of two user-defined groups. However, there is a lack of
nonparametric procedures to analyze microarray data with multiple factors
attributing to the gene expression. Furthermore, incorporating interaction
effects in the analysis of microarray data has long been of great interest
to biological scientists, little of which has been investigated in the
nonparametric framework.
Results: In this paper, we propose a set
of nonparametric tests to detect treatment effects, clinical covariate
effects, and interaction effects for multifactorial microarray data. When
the distribution of expression data is skewed or heavy-tailed, the rank
tests are substantially more powerful than the competing parametric F tests.
On the other hand, in the case of light or medium-tailed distributions,
the rank tests appear to be marginally less powerful than the parametric
competitors.
Conclusion: The proposed rank tests enable us
to detect differential gene expression and establish interaction effects
for microarray data with various non-normally distributed expression measurements
across genome. In the presence of outliers, they are advantageous alternative
approaches to the existing parametric F tests due to the robustness feature.
Yuan-De
Tan, Myriam Fornage and Hongyan Xu
Background: Microarray technology provides an
efficient means for globally exploring physiological processes governed
by the coordinated expression of multiple genes. However, identification
of genes differentially expressed in microarray experiments is challenging
because of their potentially high type I error rate. Methods for large-scale
statistical analyses have been developed but most of them are applicable
to two-sample or two-condition data.
Results: We developed a large-scale multiple-group
F-test based method, named ranking analysis of F-statistics (RAF), which
is an extension of ranking analysis of microarray data (RAM) for two sample
t-test. In this method, we proposed a novel random splitting approach to
generate the null distribution instead of using permutation, which may
not be appropriate for microarray data. We also implemented a two-simulation
strategy to estimate the false discovery rate. Simulation results suggested
that it has higher efficiency in finding differentially expressed genes
among multiple classes at a lower false discovery rate than some commonly
used methods. By applying our method to the experimental data, we found
107 genes having significantly differential expressions among 4 treatments
at <0.7% FDR, of which 31 belong to the expressed sequence tags (ESTs),
76 are unique genes who have known functions in the brain or central nervous
system and belong to six major functional groups.
Conclusion: Our method is suitable to identify
differentially expressed genes among multiple groups, in particular, when
sample size is small.
Markus
Neuhäuser and Roswitha Senske
Motivation: An important application of microarray
experiments is to identify differentially expressed genes. Because microarray
data are often not distributed according to a normal distribution nonparametric
methods were suggested for their statistical analysis. Here, the Baumgartner-Weiß-Schindler
test, a novel and powerful test based on ranks, is investigated and compared
with the parametric t -test as well as with two other nonparametric tests
(Wilcoxon rank sum test, Fisher-Pitman permutation test) recently recommended
for the analysis of gene expression data.
Results: Simulation studies show that an
exact permutation test based on the Baumgartner-Weiß-Schindler statistic
B is preferable to the other three tests. It is less conservative than
the Wilcoxon test and more powerful, in particular in case of asymmetric
or heavily tailed distributions. When the underlying distribution is symmetric
the differences in power between the tests are relatively small. Thus,
the Baumgartner-Weiß- Schindler is recommended for the usual situation
that the underlying distribution is a priori unknown.
![]()
Yingdong
Zhao, Ming-Chung Li and Richard Simon
Background: Normalization is a critical step
in analysis of gene expression profiles. For dual labeled arrays, global
normalization assumes that the majority of the genes on the array are nondifferentially
expressed between the two channels and that the number of over-expressed
genes approximately equals the number of under-expressed genes. These assumptions
can be inappropriate for custom arrays or arrays in which the reference
RNA is very different from the experimental samples.
Results: We propose a mixture model based
normalization method that adaptively identifies nondifferentially expressed
genes and thereby substantially improves normalization for dual-labeled
arrays in settings where the assumptions of global normalization are problematic.
The new method is evaluated using both simulated and real data.
Conclusions: The new normalization method is
effective for general microarray platforms when samples with very different
expression profile are co-hybridized and for custom arrays where the majority
of genes are likely to be differentially expressed.
Tim Lu, Christine
M Costello, Peter JP Croucher, Robert Häsler, Günther Deuschl and Stefan
Schreiber
Background: Normalization is the process of
removing non-biological sources of variation between array experiments.
Recent investigations of data in gene expression databases for varying
organisms and tissues have shown that the majority of expressed genes exhibit
a power-law distribution with an exponent close to -1 (i.e. obey Zipf's
law). Based on the observation that our single channel and two channel
microarray data sets also followed a power-law distribution, we were motivated
to develop a normalization method based on this law, and examine how it
compares with existing published techniques. A computationally simple and
intuitively appealing technique based on this observation is presented.
Results: Using pairwise comparisons using
MA plots (log ratio vs. log intensity), we compared this novel method to
previously published normalization techniques, namely global normalization
to the mean, the quantile method, and a variation on the loess normalization
method designed specifically for boutique microarrays. Results indicated
that, for single channel microarrays, the quantile method was superior
with regard to eliminating intensity-dependent effects (banana curves),
but Zipf's law normalization does minimize this effect by rotating the
data distribution such that the maximal number of data points lie on the
zero of the log ratio axis. For two channel boutique microarrays, the Zipf's
law normalizations performed as well as, or better than existing techniques.
Conclusion: Zipf's law normalization is a useful tool where the Quantile
method cannot be applied, as is the case with microarrays containing functionally
specific gene sets (boutique arrays).
David
C. Hoyle, Magnus Rattray, Ray Jupp and Andrew Brass
Motivation: Typical analysis of microarray data
has focused on spot by spot comparisons within a single organism. Less
analysis has been done on the comparison of the entire distribution of
spot intensities between experiments and between organisms.
Results: Here we show that mANA transcription
data from a wide range of organisms and measured with a range of experimental
platforms show close agreement with Benford's law (Benford, Proc. Am.
Phil. Soc., 78, 551-572, 1938) and Zipf's law (Zipf, The
Psycho-biology of Language: an Introduction to Dynamic Philology, 1936
and Human Behaviour and the Principle of Least Effort, 1949). The
distribution of the bulk of microarray spot intensities is weil approximated
by a log-normal with the tail of the distribution being closer to power
law. The variance, σ2, of log spot intensity shows a positive
correlation with genome size (in terms of number of genes) and is therefore
relatively fixed within some range for a given organism. The measured value
of σ2 can be significantly smaller than the expected value
if the mANA is extracted from a sample of mixed cell types. Our research
demonstrates that useful biological findings may result from analyzing
microarray data at the level of entire intensity distributions.
![]()
Radka
Stoyanova, Troy D. Querec, Truman R. Brown and Christos Patriotis
Motivation: Detailed comparison and analysis
of the output of DNA gene expression arrays from multiple samples require
global normalization of the measured individual gene intensities from the
different hybridizations. This is needed for accounting for variations
in array preparation and sample hybridization conditions.
Results: Here, we present a simple, robust
and accurate procedure for the global normalization of datasets generated
with single-channel DNAarrays based on principal component analysis. The
procedure makes minimal assumptions about the data and performs well in
cases where other standard procedures produced biased estimates. It is
also insensitive to data transformation, filtering (thresholding) and pre-screening.
Ki-Yeol
Kim, Byoung-Jin Kim and Gwan-Su Yi
Background: The imputation of missing values
is necessary for the efficient use of DNA microarray data, because many
clustering algorithms and some statistical analysis require a complete
data set. A few imputation methods for DNA microarray data have been introduced,
but the efficiency of the methods was low and the validity of imputed values
in these methods had not been fully checked.
Results: We developed a new cluster-based
imputation method called sequential K-nearest neighbor (SKNN) method. This
imputes the missing values sequentially from the gene having least missing
values, and uses the imputed values for the later imputation. Although
it uses the imputed values, the efficiency of this new method is greatly
improved in its accuracy and computational complexity over the conventional
KNN-based method and other methods based on maximum likelihood estimation.
The performance of SKNN was in particular higher than other imputation
methods for the data with high missing rates and large number of experiments.
Application
of Expectation Maximization (EM) to the SKNN method improved the accuracy,
but increased computational time proportional to the number of iterations.
The Multiple Imputation (MI) method, which is well known but not applied
previously to microarray data, showed a similarly high accuracy as the
SKNN method, with slightly higher dependency on the types of data sets.
Conclusions: Sequential reuse of imputed data
in KNN-based imputation greatly increases the efficiency of imputation.
The SKNN method should be practically useful to save the data of some microarray
experiments which have high amounts of missing entries. The SKNN method
generates reliable imputed values which can be used for further cluster-based
analysis of microarray data.
Jianqing
Fan and Yue Niu
Motivation: Normalization of microarray data
is essential for multiple-array analyses. Several normalization protocols
have been proposed based on different biological or statistical assumptions.
A fundamental problem arises whether they have effectively normalized arrays.
In addition, for a given array, the question arises how to choose a method
to most effectively normalize the microarray data.
Results: We propose several techniques to
compare the effectiveness of different normalization methods. We approach
the problem by constructing statistics to test whether there are any systematic
biases in the expression profiles among duplicated spots within an array.
The test statistics involve estimating the genewise variances. This is
accomplished by using several novel methods, including empirical Bayes
methods for moderating the genewise variances and the smoothing methods
for aggregating variance information. P-values are estimated based on a
normal or χ approximation. With estimated P-values, we can choose
a most appropriate method to normalize a specific array and assess the
extent to which the systematic biases due to the variations of experimental
conditions have been removed. The effectiveness and validity of the proposed
methods are convincingly illustrated by a carefully designed simulation
study. The method is further illustrated by an application to human placenta
cDNAs comprising a large number of clones with replications, a customized
microarray experiment carrying just a few hundred genes on the study of
the molecular roles of Interferons on tumor, and the Agilent microarrays
carrying tens of thousands of total RNA samples in the MAQC project on
the study of reproducibility, sensitivity and specificity of the data.
![]()
Tomokazu
Konishi
Background: To cancel experimental variations,
microarray data must be normalized prior to analysis. Where an appropriate
model for statistical data distribution is available, a parametric method
can normalize a group of data sets that have common distributions. Although
such models have been proposed for microarray data, they have not always
fit the distribution of real data and thus have been inappropriate for
normalization. Consequently, microarray data in most cases have been normalized
with non-parametric methods that adjust data in a pair-wise manner. However,
data analysis and the integration of resultant knowledge among experiments
have been difficult, since such normalization concepts lack a universal
standard.
Results: A three-parameter lognormal distribution
model was tested on over 300 sets of microarray data. The model treats
the hybridization background, which is difficult to identify from images
of hybridization, as one of the parameters. A rigorous coincidence of the
model to data sets was found, proving the model's appropriateness for microarray
data. In fact, a closer fitting to Northern analysis was obtained. The
model showed inconsistency only at very strong or weak data intensities.
Measurement of z-scores as well as calculated ratios was reproducible only
among data in the model-consistent intensity range; also, the ratios were
independent of signal intensity at the corresponding range.
Conclusion: The model could provide a universal
standard for data, simplifying data analysis and knowledge integration.
It was deduced that the ranges of inconsistency were caused by experimental
errors or additive noise in the data; therefore, excluding the data corresponding
to those marginal ranges will prevent misleading analytical conclusions.
Huiling
Xiong, Dapeng Zhang, Christopher J Martyniuk, Vance L Trudeau and Xuhua
Xia
Background: Normalization is essential in dual-labelled
microarray data analysis to remove nonbiological variations and systematic
biases. Many normalization methods have been used to remove such biases
within slides (Global, Lowess) and across slides (Scale, Quantile and VSN).
However, all these popular approaches have critical assumptions about data
distribution, which is often not valid in practice.
Results: In this study, we propose a novel
assumption-free normalization method based on the Generalized Procrustes
Analysis (GPA) algorithm. Using experimental and simulated normal microarray
data and boutique array data, we systemically evaluate the ability of the
GPA method in normalization compared with six other popular normalization
methods including Global, Lowess, Scale, Quantile, VSN, and one boutique
array-specific housekeeping gene method. The assessment of these methods
is based on three different empirical criteria: across-slide variability,
the Kolmogorov-Smirnov (K-S) statistic and the mean square error (MSE).
Compared with other methods, the GPA method performs effectively and consistently
better in reducing across-slide variability and removing systematic bias.
Conclusion: The GPA method is an effective normalization
approach for microarray data analysis. In particular, it is free from the
statistical and biological assumptions inherent in other normalization
methods that are often difficult to validate. Therefore, the GPA method
has a major advantage in that it can be applied to diverse types of array
sets, especially to the boutique array where the majority of genes may
be differentially expressed.
Tristan
Mary-Huard, Jean-Jacques Daudin, Michela Baccini, Annibale Biggeri and
Avner Bar-Hen
Motivation: If there is insufficient RNA from
the tissues under investigation from one organism, then it is common practice
to pool RNA. An important question is to determine whether pooling introduces
biases, which can lead to inaccurate results. In this article, we describe
two biases related to pooling, from a theoretical as well as a practical
point of view.
Results: We model and quantify the respective
parts of the pooling bias due to the log transform as well as the bias
due to biological averaging of the samples. We also evaluate the impact
of the bias on the statistical differential analysis of Affymetrix data.
![]()
Shu-Dong
Zhang and Timothy W. Gant
Motivation: Many biomedical experiments are
carried out by pooling individual biological samples. However, pooling
samples can potentially hide biological variance and give false confidence
concerning the data significance. In the context of microarray experiments
for detecting differentially expressed genes, recent publications have
addressed the problem of the efficiency of sample pooling, and some approximate
formulas were provided for the power and sample size calculations. It is
desirable to have exact formulas for these calculations and have the approximate
results checked against the exact ones. We show that the difference between
the approximate and the exact results can be large.
Results: In this study, we have characterized
quantitatively the effect of pooling samples on the efficiency of microarray
experiments for the detection of differential gene expression between two
classes. We present exact formulas for calculating the power of microarray
experimental designs involving sample pooling and technical replications.
The formulas can be used to determine the total number of arrays and biological
subjects required in an experiment to achieve the desired power at a given
significance level. The conditions under which pooled design becomes preferable
to non-pooled design can then be derived given the unit cost associated
with a microarray and that with a biological subject. This paper thus serves
to provide guidance on sample pooling and cost-effectiveness. The formulation
in this paper is outlined in the context of performing microarray comparative
studies, but its applicability is not limited to microarray experiments.
It is also applicable to a wide range of biomedical comparative studies
where sample pooling may be involved.
Wuyan
Zhang, Alicia Carriquiry, Dan Nettleton and Jack C.M. Dekkers
Motivation: Microarrays can simultaneously measure
the expression levels of many genes and are widely applied to study complex
biological problems at the genetic level. To contain costs, instead of
obtaining a microarray on each individual, mRNA from several subjects can
be first pooled and then measured with a single array. mRNA pooling is
also necessary when there is not enough mRNA from each subject. Several
studies have investigated the impact of pooling mRNA on inferences about
gene expression, but have typically modeled the process of pooling as if
it occurred in some transformed scale. This assumption is unrealistic.
Results: We propose modeling the gene expression
levels in a pool as a weighted average of mRNA expression of all individuals
in the pool on the original measurement scale, where the weights correspond
to individual sample contributions to the pool. Based on these improved
statistical models, we develop the appropriate F statistics to test for
differentially expressed genes. We present formulae to calculate the power
of various statistical tests under different strategies for pooling mRNA
and compare resulting power estimates to those that would be obtained by
following the approach proposed by Kendziorski et al. (2003). We find that
the Kendziorski estimate tends to exceed true power and that the estimate
we propose, while somewhat conservative, is less biased. We argue that
it is possible to design a study that includes mRNA pooling at a significantly
reduced cost but with little loss of information.
Xuejun
Peng, Constance L Wood, Eric M Blalock, Kuey Chu Chen, Philip W Landfield
and Arnold J Stromberg
Background: Microarray technology has become
a very important tool for studying gene expression profiles under various
conditions. Biologists often pool RNA samples extracted from different
subjects onto a single microarray chip to help defray the cost of microarray
experiments as well as to correct for the technical difficulty in getting
sufficient RNA from a single subject. However, the statistical, technical
and financial implications of pooling have not been explicitly investigated.
Results: Modeling the resulting gene expression
from sample pooling as a mixture of individual responses, we derived expressions
for the experimental error and provided both upper and lower bounds for
its value in terms of the variability among individuals and the number
of RNA samples pooled. Using "virtual" pooling of data from real
experiments and computer simulations, we investigated the statistical properties
of RNA sample pooling. Our study reveals that pooling biological samples
appropriately is statistically valid and efficient for microarray experiments.
Furthermore, optimal pooling design(s) can be found to meet statistical
requirements while minimizing total cost.
Conclusions: Appropriate RNA pooling can provide
equivalent power and improve efficiency and cost-effectiveness for microarray
experiments with a modest increase in total number of subjects. Pooling
schemes in terms of replicates of subjects and arrays can be compared before
experiments are conducted.
![]()
Shigeyuki
Oba, Masa-aki Sato, Ichiro Takemasa, Morito Monden, Ken-ichi Matsubara
and Shin Ishii
Motivation: Gene expression profile analyses
have been used in numerous studies covering a broad range of areas in biology.
When unreliable measurements are excluded, missing values are introduced
in gene expression profiles. Although existing multivariate analysis methods
have difficulty with the treatment of missing values, this problem has
received little attention. There are many options for dealing with missing
values, each of which reaches drastically different results. Ignoring missing
values is the simplest method and is frequently applied. This approach,
however, has its flaws. In this article, we propose an estimation method
for missing values, which is based on Bayesian principal component analysis
(BPCA). Although the methodology that a probabilistic model and latent
variables are estimated simultaneously within the framework of Bayes inference
is not new in principle, actual BPCA implementation that makes it possible
to estimate arbitrary missing variables is new in terms of statistical
methodology.
Results: When applied to DNA microarray data
from various experimental conditions, theBPCAmethod exhibited markedly
better estimation ability than other recently proposed methods, such as
singular value decomposition and K -nearest neighbors. While the estimation
performance of existing methods depends on model parameters whose determination
is difficult, our BPCA method is free from this difficulty. Accordingly,
the BPCA method provides accurate and convenient estimation for missing
values.
Matthew
E. Ritchie, Jeremy Silver, Alicia Oshlack, Melissa Holmes, Dileepa Diyagama,
Andrew Holloway and Gordon K. Smyth
Motivation: Microarray data must be background
corrected to remove the effects of non-specific binding or spatial heterogeneity
across the array, but this practice typically causes other problems such
as negative corrected intensities and high variability of low intensity
log-ratios. Different estimators of background, and various model-based
processing methods, are compared in this study in search of the best option
for differential expression analyses of small microarray experiments.
Results: Using data where some independent
truth in gene expression is known, eight different background correction
alternatives are compared, in terms of precision and bias of the resulting
gene expression measures, and in terms of their ability to detect differentially
expressed genes as judged by two popular algorithms, SAM and limma eBayes.
A new background processing method (normexp) is introduced which is based
on a convolution model. The model-based correction methods are shown to
be markedly superior to the usual practice of subtracting local background
estimates. Methods which stabilize the variances of the log-ratios along
the intensity range perform the best. The normexp+offset method is found
to give the lowest false discovery rate overall, followed by morph and
vsn. Like vsn, normexp is applicable to most types of two-colour microarray
data.
Troy
D. Querec, Radka Stoyanova, Eric Ross and Christos Patriotis
Motivation: The radioactivity labeled DNA array
platform is a robust and accurate way for a high-throughput measurement
of gene expression levels in biological samples. Despite its high degree
of sensitivity and reproducibility, this platform has several sources of
variation.These are related to the presence of saturation effects in the
array images and impede the degree of accuracy at which gene expression
levels are determined.
Results: Here we describe a simple, but effective,
approach for combining expression data from a series of autoradiographic
exposures of variable length. This technique increases the sensitivity
of this array platformby detecting low-expressed genes at longer exposures.
It also improves the measurement accuracy of highly abundant genes by considering
only values from the linear portion of dependency between the exposure
times and gene intensities.As a result, the described approach improves
the outcome of the subsequent steps of array data normalization and mining.
![]()
H. P.
Piepho, B. Keller, N. Hoecker and F. Hochholdinger
Motivation: The analysis of spotted cDNA microarrays
involves scanning of color signals from fluorescent dyes. A common problem
is that a given scanning intensity is not usually optimal for all spotted
cDNAs. Specifically, some spots may be at the saturation limit, resulting
in poor separation of signals from different tissues or conditions. The
problem may be addressed by multiple scans with varying scanning intensities.
Multiple scanning intensities raise the question of how to combine different
signals from the same spot, particularly when measurement error is not
negligible.
Results: This paper suggests a non-linear
latent regression model for this purpose. It corrects for biases caused
by the saturation limit and efficiently combines data from multiple scans.
Combining multiple scans also allows reduction of technical error particularly
for cDNA spots with low signal. The procedure is exemplified using cDNA
expression data from maize.
Helene H Thygesen
and Aeilko H Zwinderman
Background: When DNA microarray data are used
for gene clustering, genotype/phenotype correlation studies, or tissue
classification the signal intensities are usually transformed and normalized
in several steps in order to improve comparability and signal/noise ratio.
These steps may include subtraction of an estimated background signal,
subtracting the reference signal, smoothing (to account for nonlinear measurement
effects), and more. Different authors use different approaches, and it
is generally not clear to users which method they should prefer.
Results: We used the ratio between biological
variance and measurement variance (which is an Flike statistic) as a quality
measure for transformation methods, and we demonstrate a method for maximizing
that variance ratio on real data. We explore a number of transformations
issues, including Box-Cox transformation, baseline shift, partial subtraction
of the log-reference signal and smoothing. It appears that the optimal
choice of parameters for the transformation methods depends on the data.
Further, the behavior of the variance ratio, under the null hypothesis
of zero biological variance, appears to depend on the choice of parameters.
Conclusions: The use of replicates in microarray
experiments is important. Adjustment for the null-hypothesis behavior of
the variance ratio is critical to the selection of transformation method.
Ryan
Kelley, Hoda Feizi and Trey Ideker
Motivation: In two-color microarray experiments,
well-known differences exist in the labeling and hybridization efficiency
of Cy3 and Cy5 dyes. Previous reports have revealed that these differences
can vary on a gene-by-gene basis, an effect termed gene-specific dye bias.
If uncorrected, this bias can influence the determination of differentially
expressed genes.
Results: We show that the magnitude of the
bias scales multiplicatively with signal intensity and is dependent on
which nucleotide has been conjugated to the fluorescent dye. A method is
proposed to account for gene-specific dye bias within a maximum-likelihood
error modeling framework. Using two different labeling schemes, we show
that correcting for gene-specific dye bias results in the superior identification
of differentially expressed genes within this framework. Improvement is
also possible in related ANOVA approaches.
Ming
Ouyang, William J. Welsh and Panos Georgopoulos
Motivation: In microarray experiments, missing
entries arise from blemishes on the chips. In large-scale studies, virtually
every chip contains some missing entries and more than 90% of the genes
are affected. Many analysis methods require a full set of data. Either
those genes with missing entries are excluded, or the missing entries are
filled with estimates prior to the analyses.This study compares methods
of missing value estimation.
Results: Two evaluation metrics of imputation
accuracy are employed. First, the root mean squared error measures the
difference between the true values and the imputed values. Second, the
number of mis-clustered genes measures the difference between clustering
with true values and that with imputed values; it examines the bias introduced
by imputation to clustering. The Gaussian mixture clustering with model
averaging imputation is superior to all other imputation methods, according
to both evaluation metrics, on both time-series (correlated) and non-time
series (uncorrelated) data sets.
![]()
Anders
Bengtsson and Henrik Bengtsson
Background: In a microarray experiment the difference
in expression between genes on the same slide is up to 103 fold
or more. At low expression, even a small error in the estimate will have
great influence on the final test and reference ratios. In addition to
the true spot intensity the scanned signal consists of different kinds
of noise referred to as background. In order to assess the true spot intensity
background must be subtracted. The standard approach to estimate background
intensities is to assume they are equal to the intensity levels between
spots. In the literature, morphological opening is suggested to be one
of the best methods for estimating background this way.
Results: This paper examines fundamental
properties of rank and quantile filters, which include morphological filters
at the extremes, with focus on their ability to estimate between-spot intensity
levels. The bias and variance of these filter estimates are driven by the
number of background pixels used and their distributions. A new rank-filter
algorithm is implemented and compared to methods available in Spot by CSIRO
and GenePix Pro by Axon Instruments. Spot's morphological opening has a
mean bias between -47 and -248 compared to a bias between 2 and -2 for
the rank filter and the variability of the morphological opening estimate
is 3 times higher than for the rank filter. The mean bias of Spot's second
method, morph.close.open, is between -5 and -16 and the variability is
approximately the same as for morphological opening. The variability of
GenePix Pro's region based estimate is more than ten times higher than
the variability of the rank-filter estimate and with slightly more bias.
The large variability is because the size of the background window changes
with spot size. To overcome this, a non-adaptive region-based method is
implemented. Its bias and variability are comparable to that of the rank
filter.
Conclusion: The performance of more advanced
rank filters is equal to the best region-based methods. However, in order
to get unbiased estimates these filters have to be implemented with great
care. The performance of morphological opening is in general poor with
a substantial spatial dependent bias.
Xiaobo
Zhou, Xiaodong Wang and Edward R. Dougherty
Motivation: Data from microarray experiments
are usually in the form of large matrices of expression levels of genes
under different experimental conditions. Owing to various reasons, there
are frequently missing values. Estimating these missing values is important
because they affect downstream analysis, such as clustering, classification
and network design. Several methods of missing-value estimation are in
use. The problem has two parts: (1) selection of genes for estimation and
(2) design of an estimation rule.
Results: We propose Bayesian variable selection
to obtain genes to be used for estimation, and employ both linear and nonlinear
regression for the estimation rule itself. Fast implementation issues for
these methods are discussed, including the use of QR decomposition for
parameter estimation. The proposed methods are tested on data sets arising
from hereditary breast cancer and small round blue-cell tumors.The results
compare very favorably with currently used methods based on the normalized
root-mean-square error.
Mark
Reimers and John N Weinstein
Background: Quality-control is an important
issue in the analysis of gene expression microarrays. One type of problem
is regional bias, in which one region of a chip shows artifactually high
or low intensities (or ratios in a two-channel array) relative to the majority
of the chip. Current practice in quality assessment for microarrays does
not address regional biases.
Results: We present methods implemented in
R for visualizing regional biases and other spatial artifacts on spotted
microarrays and Affymetrix chips. We also propose a statistical index to
quantify regional bias and investigate its typical distribution on spotted
and Affymetrix arrays. We demonstrate that notable regional biases occur
on both Affymetrix and spotted arrays and that they can make a significant
difference in the case of spotted microarray results. Although strong biases
are also seen at the level of individual probes on Affymetrix chips, the
gene expression measures are less affected, especially when the RMA method
is used to summarize intensities for the probe sets. A web application
program for visualization and quantitation of regional bias is provided
at http://www.discover.nci.nih.gov/affytools.
Conclusion: Researchers should visualize and
measure the regional biases and should estimate their impact on gene expression
measurements obtained. Here, we (i) introduce pictorial visualizations
of the spatial biases; (ii) present for Affymetrix chips a useful resolution
of the biases into two components, one related to background, the other
to intensity scale factor; (iii) introduce a single parameter to reflect
the global bias present across an array. We also examine the pattern distribution
of such biases and conclude that algorithms based on smoothing are unlikely
to compensate adequately for them.
![]()
Brian
DM Tom, Walter R Gilks, Elizabeth T Brooke-Powell and James W Ajioka
Background: A common feature of microarray experiments
is the occurence of missing gene expression data. These missing values
occur for a variety of reasons, in particular, because of the filtering
of poor quality spots and the removal of undefined values when a logarithmic
transformation is applied to negative background-corrected intensities.
The efficiency and power of an analysis performed can be substantially
reduced by having an incomplete matrix of gene intensities. Additionally,
most statistical methods require a complete intensity matrix. Furthermore,
biases may be introduced into analyses through missing information on some
genes. Thus methods for appropriately replacing (imputing) missing data
and/or weighting poor quality spots are required.
Results: We present a likelihood-based method
for imputing missing data or weighting poor quality spots that requires
a number of biological or technical replicates. This likelihood-based approach
assumes that the data for a given spot arising from each channel of a two-dye
(two channel) cDNA microarray comparison experiment independently come
from a three-component mixture distribution the parameters of which are
estimated through use of a constrained E-M algorithm. Posterior probabilities
of belonging to each component of the mixture distributions are calculated
and used to decide whether imputation is required. These posterior probabilities
may also be used to construct quality weights that can down-weight poor
quality spots in any analysis performed afterwards. The approach is illustrated
using data obtained from an experiment to observe gene expression changes
with 24 hr paclitaxel (Taxol ®) treatment on a human cervical cancer derived
cell line (HeLa).
Conclusion: As the quality of microarray experiments
affect downstream processes, it is important to have a reliable and automatic
method of identifying poor quality spots and arrays. We propose a method
of identifying poor quality spots, and suggest a method of repairing the
arrays by either imputation or assigning quality weights to the spots.
This repaired data set would be less biased and can be analysed using any
of the appropriate statistical methods found in the microarray literature.
Heidi
Lyng, Azadeh Badiee, Debbie H Svendsrud, Eivind Hovig, Ola Myklebost and
Trond Stokke
Background: High throughput gene expression
data from spotted cDNA microarrays are collected by scanning the signal
intensities of the corresponding spots by dedicated fluorescence scanners.
The major scanner settings for increasing the spot intensities are the
laser power and the voltage of the photomultiplier tube (PMT). It is required
that the expression ratios are independent of these settings. We have investigated
the relationships between PMT voltage, spot intensities, and expression
ratios for different scanners, in order to define an optimal scanning procedure.
Results: All scanners showed a limited intensity
range from 200 to 50 000 (mean spot intensity), for which the expression
ratios were independent of PMT voltage. This usable intensity range was
considerably less than the maximum detection range of the PMTs. The use
of spot and background intensities outside this range led to errors in
the ratios. The errors at high intensities were caused by saturation of
pixel intensities within the spots. An algorithm was developed to correct
the intensities of these spots, and, hence, extend the upper limit of the
usable intensity range.
Conclusions: It is suggested that the PMT voltage
should be increased to avoid intensities of the weakest spots below the
usable range, allowing the brightest spots to reach the level of saturation.
Subsequently, a second set of images should be acquired with a lower PMT
setting such that no pixels are in saturation. Reliable data for spots
with saturation in the first set of images can easily be extracted from
the second set of images by the use of our algorithm. This procedure would
lead to an increase in the accuracy of the data and in the number of data
points achieved in each experiment compared to traditional procedures.
David
S. Skibbe, Xiujuan Wang, Xuefeng Zhao, Lisa A. Borsuk, Dan Nettleton and
Patrick S. Schnable
Motivation: Scanning parameters are often overlooked
when optimizing microarray experiments. A scanning approach that extends
the dynamic data range by acquiring multiple scans of different intensities
has been developed.
Results: Data from each of three scan intensities
(low, medium, high) were analyzed separately using multiple scan and linear
regression approaches to identify and compare the sets of genes that exhibit
statistically significant differential expression. In the multiple scan
approach only one-third of the differentially expressed genes were shared
among the three intensities, and each scan intensity identified unique
sets of differentially expressed genes. The set of differentially expressed
genes from any one scan amounted to <70% of the total number of genes
identified in at least one scan. The average signal intensity of genes
that exhibited statistically significant changes in expression was highest
for the low-intensity scan and lowest for the high-intensity scan, suggesting
that low-intensity scans may be best for detecting expression differences
in high-signal genes, while high-intensity scans may be best for detecting
expression differences in low-signal genes. Comparison of the differentially
expressed genes identified in the multiple scan and linear regression approaches
revealed that the multiple scan approach effectively identifies a subset
of statistically significant genes that linear regression approach is unable
to identify. Quantitative RTPCR (qRTPCR) tests demonstrated that statistically
significant differences identified at all three scan intensities can be
verified.
![]()
Mizanur
R. Khondoker, Chris A. Glasbey and Bruce J. Worton
We propose
a statistical model for estimating gene expression using data from multiple
laser scans at different settings of hybridized microarrays. A functional
regression model is used, based on a non-linear relationship with both
additive and multiplicative error terms. The function is derived as the
expected value of a pixel, given that values are censored at 65 535, the
maximum detectable intensity for double precision scanning software. Maximum
likelihood estimation based on a Cauchy distribution is used to fit the
model, which is able to estimate gene expressions taking account of outliers
and the systematic bias caused by signal censoring of highly expressed
genes. We have applied the method to experimental data. Simulation studies
suggest that the model can estimate the true gene expression with negligible
bias.
Mariano
J Alvarez, Guillermo J Vila-Ortiz, Mariano C Salibe, Osvaldo L Podhajcer
and Fernando J Pitossi
Background: Reverse transcription followed by
real-time PCR is widely used for quantification of specific mRNA, and with
the use of double-stranded DNA binding dyes it is becoming a standard for
microarray data validation. Despite the kinetic information generated by
real-time PCR, most popular analysis methods assume constant amplification
efficiency among samples, introducing strong biases when amplification
efficiencies are not the same.
Results: We present here a new mathematical
model based on the classic exponential description of the PCR, but modeling
amplification efficiency as a sigmoidal function of the product yield.
The model was validated with experimental results and used for the development
of a new method for real-time PCR data analysis. This model based method
for real-time PCR data analysis showed the best accuracy and precision
compared with previous methods when used for quantification of in silico
generated and experimental real-time PCR results. Moreover, the method
is suitable for the analyses of samples with similar or dissimilar amplification
efficiency.
Conclusion: The presented method showed the
best accuracy and precision. Moreover, it does not depend on calibration
curves, making it ideal for fully automated high-throughput applications.
Anke
Batsch, Andrea Noetel, Christian Fork, Anita Urban, Daliborka Lazic, Tina
Lucas, Julia Pietsch, Andreas Lazar, Edgar Schömig and Dirk Gründemann
Background: In real-time PCR, it is necessary
to consider the efficiency of amplification (EA) of amplicons in order
to determine initial target levels properly. EAs can be deduced from standard
curves, but these involve extra effort and cost and may yield invalid EAs.
Alternatively, EA can be extracted from individual fluorescence curves.
Unfortunately, this is not reliable enough.
Results: Here we introduce simultaneous non-linear
fitting to determine without standard curves an optimal common EA for
all samples of a group. In order to adjust EA as a function of target fluorescence,
and still to describe fluorescence as a function of cycle number, we use
an iterative algorithm that increases fluorescence cycle by cycle and thus
simulates the PCR process. A Gauss peak function is used to model the decrease
of EA with increasing amplicon accumulation. Our approach was validated
experimentally with hydrolysis probe or SYBR green detection with dilution
series of 5 different targets. It performed distinctly better in terms
of accuracy than standard curve, DART-PCR, and LinRegPCR approaches. Based
on reliable EAs, it was possible to detect that for some amplicons, extraordinary
fluorescence (EA > 2.00) was generated with locked nucleic acid hydrolysis
probes, but not with SYBR green.
Conclusion: In comparison to previously reported
approaches that are based on the separate analysis of each curve and on
modelling EA as a function of cycle number, our approach yields more accurate
and precise estimates of relative initial target levels.
Joshua
S Yuan, Ann Reed, Feng Chen and C Neal Stewart Jr
Background: Even though real-time PCR has been
broadly applied in biomedical sciences, data processing procedures for
the analysis of quantitative real-time PCR are still lacking; specifically
in the realm of appropriate statistical treatment. Confidence interval
and statistical significance considerations are not explicit in many of
the current data analysis approaches. Based on the standard curve method
and other useful data analysis methods, we present and compare four statistical
approaches and models for the analysis of real-time PCR data.
Results: In the first approach, a multiple
regression analysis model was developed to derive ΔΔCt from estimation
of interaction of gene and treatment effects. In the second approach, an
ANCOVA (analysis of covariance) model was proposed, and the ΔΔCt
can be derived from analysis of effects of variables. The other two models
involve calculation ΔCt followed by a two group t-test and nonparametric
analogous Wilcoxon test. SAS programs were developed for all four models
and data output for analysis of a sample set are presented. In addition,
a data quality control model was developed and implemented using SAS.
Conclusion: Practical statistical solutions
with SAS programs were developed for real-time PCR data and a sample dataset
was analyzed with the SAS programs. The analysis using the various models
and programs yielded similar results. Data quality control and analysis
procedures presented here provide statistical elements for the estimation
of the relative expression of genes using real-time PCR.
![]()
Yann
Karlen, Alan McNair, Sébastien Perseguers, Christian Mazza and Nicolas
Mermod
Background: PCR has the potential to detect
and precisely quantify specific DNA sequences, but it is not yet often
used as a fully quantitative method. A number of data collection and processing
strategies have been described for the implementation of quantitative PCR.
However, they can be experimentally cumbersome, their relative performances
have not been evaluated systematically, and they often remain poorly validated
statistically and/or experimentally. In this study, we evaluated the performance
of known methods, and compared them with newly developed data processing
strategies in terms of resolution, precision and robustness.
Results: Our results indicate that simple
methods that do not rely on the estimation of the efficiency of the PCR
amplification may provide reproducible and sensitive data, but that they
do not quantify DNA with precision. Other evaluated methods based on sigmoidal
or exponential curve fitting were generally of both poor resolution and
precision. A statistical analysis of the parameters that influence efficiency
indicated that it depends mostly on the selected amplicon and to a lesser
extent on the particular biological sample analyzed. Thus, we devised various
strategies based on individual or averaged efficiency values, which were
used to assess the regulated expression of several genes in response to
a growth factor.
Conclusion: Overall, qPCR data analysis methods
differ significantly in their performance, and this analysis identifies
methods that provide DNA quantification estimates of high precision, robustness
and reliability. These methods allow reliable estimations of relative expression
ratio of two-fold or higher, and our analysis provides an estimation of
the number of biological samples that have to be analyzed to achieve a
given precision.
Ziv Bar-Joseph
Motivation: Time series expression experiments
are an increasingly popular method for studying a wide range of biological
systems. However, when analyzing these experiments researchers face many
new computational challenges. Algorithms that are specifically designed
for time series experiments are required so that we can take advantage
of their unique features (such as the ability to infer causality from the
temporal response pattern) and address the unique problems they raise (e.g.
handling the different non-uniform sampling rates).
Results: We present a comprehensive review
of the current research in time series expression data analysis. We divide
the computational challenges into four analysis levels: experimental design,
data analysis, pattern recognition and networks. For each of these levels,
we discuss computational and biological problems at that level and point
out some of the methods that have been proposed to deal with these issues.
Many open problems in all these levels are discussed. This review is intended
to serve as both, a point of reference for experimental biologists looking
for practical solutions for analyzing their data, and a starting point
for computer scientists interested in working on the computational problems
related to time series expression analysis.
Matthias
E. Futschik and Hanspeter Herzel
Motivation: Periodic processes play fundamental
roles in organisms. Prominent examples are the cell cycle and the circadian
clock. Microarray array technology has enabled us to screen complete sets
of transcripts for possible association with such fundamental periodic
processes on a system-wide level. Frequently, quite large numbers of genes
have been detected as periodically expressed. However, the small overlap
between genes identified in different studies has cast some doubts on the
reliability of the periodic expression detected.
Results: In this study, comparative analysis
suggests that the lacking agreement between different cell-cycle studies
might be due to inadequate background models for the determination of significance.
We demonstrate that the choice of background model has considerable impact
on the statistical significance of periodic expression. For illustration,
we reanalyzed two microarray studies of the yeast cell cycle. Our evaluation
strongly indicates that the results of previous analyses might have been
overoptimistic and that the use of more suitable background model promises
to give more realistic results.
![]()
Jihoon
Kim and Ju Han Kim
Background: There are some limitations associated
with conventional clustering methods for short time-course gene expression
data. The current algorithms require prior domain knowledge and do not
incorporate information from replicates. Moreover, the results are not
always easy to interpret biologically.
Results: We propose a novel algorithm for
identifying a subset of genes sharing a significant temporal expression
pattern when replicates are used. Our algorithm requires no prior knowledge,
instead relying on an observed statistic which is based on the first and
second order differences between adjacent time-points. Here, a pattern
is predefined as the sequence of symbols indicating direction and the rate
of change between time-points, and each gene is assigned to a cluster whose
members share a similar pattern. We evaluated the performance of our algorithm
to those of K-means, Self-Organizing Map and the Short Time-series Expression
Miner methods.
Conclusions: Assessments using simulated and
real data show that our method outperformed aforementioned algorithms.
Our approach is an appropriate solution for clustering short timecourse
microarray data with replicates.
Neal
S. Holter, Madhusmita Mitra, Amos Maritan, Marek Cieplak, Jayanth R. Banavar
and Nina V. Fedoroff
Analysis
of previously published sets of DNA microarray gene expression data by
singular value decomposition has uncovered underlying patterns or characteristic
modes in their temporal profiles. These patterns contribute unequally
to the structure of the expression profiles. Moreover, the essential features
of a given set of expression profiles are captured using just a small number
of characteristic modes. This leads to the striking conclusion that the
transcriptional response of a genome is orchestrated in a few fundamental
patterns of gene expression change. These patterns are both simple and
robust, dominating the alterations in expression of genes throughout the
genome. Moreover, the characteristic modes of gene expression change in
response to environmental perturbations are similar in such distant organisms
as yeast and human cells. This analysis reveals simple regularities in
the seemingly complex transcriptional transitions of diverse cells to new
states, and these provide insights into the operation of the underlying
genetic networks.
Hao Li,
Constance L Wood, Yushu Liu, Thomas V Getchell, Marilyn L Getchell, and
Arnold J Stromberg
Background: In gene networks, the timing of
significant changes in the expression level of each gene may be the most
critical information in time course expression profiles. With the same
timing of the initial change, genes which share similar patterns of expression
for any number of sampling intervals from the beginning should be considered
co-expressed at certain level(s) in the gene networks. In addition, multiple
testing problems are complicated in experiments with multi-level treatments
when thousands of genes are involved.
Results: To address these issues, we first
performed an ANOVA F test to identify significantly regulated genes. The
Benjamini and Hochberg (BH) procedure of controlling false discovery rate
(FDR) at 5% was applied to the P values of the F test. We then categorized
the genes with a significant F test into 4 classes based on the timing
of their initial responses by sequentially testing a complete set of orthogonal
contrasts, the reverse Helmert series. For genes within each class, specific
sequences of contrasts were performed to characterize their general 'fluctuation'
shapes of expression along the subsequent sampling time points. To be consistent
with the BH procedure, each contrast was examined using a stepwise Studentized
Maximum Modulus test to control the gene based maximum family-wise error
rate (MFWER) at the level of a new determined by the BH procedure. We demonstrated
our method on the analysis of microarray data from murine olfactory sensory
epithelia at five different time points after target ablation.
Conclusion: In this manuscript, we used planned
linear contrasts to analyze time-course microarray experiments. This analysis
allowed us to characterize gene expression patterns based on the temporal
order in the data, the timing of a gene's initial response, and the general
shapes of gene expression patterns along the subsequent sampling time points.
Our method is particularly suitable for analysis of microarray experiments
in which it is often difficult to take sufficiently frequent measurements
and/or the sampling intervals are non-uniform.
![]()
Feng
He and An-Ping Zeng
Background: The increasing availability of time-series
expression data opens up new possibilities to study functional linkages
of genes. Present methods used to infer functional linkages between genes
from expression data are mainly based on a point-to-point comparison. Change
trends between consecutive time points in time-series data have been so
far not well explored.
Results: In this work we present a new method
based on extracting main features of the change trend and level of gene
expression between consecutive time points. The method, termed as trend
correlation (TC), includes two major steps: 1, calculating a maximal local
alignment of change trend score by dynamic programming and a change trend
correlation coefficient between the maximal matched change levels of each
gene pair; 2, inferring relationships of gene pairs based on two statistical
extraction procedures. The new method considers time shifts and inverted
relationships in a similar way as the local clustering (LC) method but
the latter is merely based on a point-to-point comparison. The TC method
is demonstrated with data from yeast cell cycle and compared with the LC
method and the widely used Pearson correlation coefficient (PCC) based
clustering method. The biological significance of the gene pairs is examined
with several large-scale yeast databases. Although the TC method predicts
an overall lower number of gene pairs than the other two methods at a same
p-value threshold, the additional number of gene pairs inferred by the
TC method is considerable: e.g. 20.5% compared with the LC method and 49.6%
with the PCC method for a p-value threshold of 2.7E-3. Moreover, the percentage
of the inferred gene pairs consistent with databases by our method is generally
higher than the LC method and similar to the PCC method. A significant
number of the gene pairs only inferred by the TC method are process-identity
or functionsimilarity pairs or have well-documented biological interactions,
including 443 known protein interactions and some known cell cycle related
regulatory interactions. It should be emphasized that the overlapping of
gene pairs detected by the three methods is normally not very high, indicating
a necessity of combining the different methods in search of functional
association of genes from time-series data. For a p-value threshold of
1E-5 the percentage of process-identity and function-similarity gene pairs
among the shared part of the three methods reaches 60.2% and 55.6% respectively,
building a good basis for further experimental and functional study. Furthermore,
the combined use of methods is important to infer more complete regulatory
circuits and network as exemplified in this study.
Conclusion: The TC method can significantly
augment the current major methods to infer functional linkages and biological
network and is well suitable for exploring temporal relationships of gene
expression in time-series data.
Eduardo
Sontag, Anatoly Kiyatkin and Boris N. Kholodenko
Motivation: High-throughput technologies have
facilitated the acquisition of large genomics and proteomics datasets.
However, these data provide snapshots of cellular behavior, rather than
help us reveal causal relations. Here, we propose how these technologies
can be utilized to infer the topology and strengths of connections among
genes, proteins and metabolites by monitoring time-dependent responses
of cellular networks to experimental interventions.
Results: We demonstrate that all connections
leading to a given network node, e.g. to a particular gene, can be deduced
from responses to perturbations none of which directly influences that
node, e.g. using strains with knock-outs to other genes. To infer all interactions
from stationary data, each node should be perturbed separately or in combination
with other nodes. Monitoring time series provides richer information and
does not require perturbations to all nodes. Overall, the methods we propose
are capable of deducing and quantifying functional interactions within
and across cellular gene, signaling and metabolic networks.
Andrey
A Ptitsyn, Sanjin Zvonic and Jeffrey M Gimble
Background: Periodic processes, such as the
circadian rhythm, are important factors modulating and coordinating transcription
of genes governing key metabolic pathways. Theoretically, even small fluctuations
in the orchestration of circadian gene expression patterns among different
tissues may result in functional asynchrony at the organism level and may
contribute to a wide range of pathologic disorders. Identification of circadian
expression pattern in time series data is important, but equally challenging.
Microarray technology allows estimation of relative expression of thousands
of genes at each time point. However, this estimation often lacks precision
and microarray experiments are prohibitively expensive, limiting the number
of data points in a time series expression profile. The data produced in
these experiments carries a high degree of stochastic variation, obscuring
the periodic pattern and a limited number of replicates, typically covering
not more than two complete periods of oscillation.
Results: To address this issue, we have developed
a simple, but effective, computational technique for the identification
of a periodic pattern in relatively short time series, typical for microarray
studies of circadian expression. This test is based on a random permutation
of time points in order to estimate non-randomness of a periodogram. The
Permutated time, or Pt-test, is able to detect oscillations within a given
period in expression profiles dominated by a high degree of stochastic
fluctuations or oscillations of different irrelevant frequencies. We have
conducted a comprehensive study of circadian expression on a large data
set produced at PBRC, representing three different peripheral murine tissues.
We have also re-analyzed a number of similar time series data sets produced
and published independently by other research groups over the past few
years.
Conclusion: The Permutated time test (Pt-test)
is demonstrated to be effective for detection of periodicity in short time
series typical for high-density microarray experiments. The software is
a set of C++ programs available from the authors on the open source basis.
![]()
Miika
Ahdesmäki, Harri Lähdesmäki, Andrew Gracey, llya Shmulevich and Olli Yli-Harja
Background: In practice many biological time
series measurements, including gene microarrays, are conducted at time
points that seem to be interesting in the biologist's opinion and not necessarily
at fixed time intervals. In many circumstances we are interested in finding
targets that are expressed periodically. To tackle the problems of uneven
sampling and unknown type of noise in periodicity detection, we propose
to use robust regression.
Methods: The aim of this paper is to develop
a general framework for robust periodicity detection and review and rank
different approaches by means of simulations. We also show the results
for some real measurement data.
Results: The simulation results clearly show
that when the sampling of time series gets more and more uneven, the methods
that assume even sampling become unusable. We find that M-estimation provides
a good compromise between robustness and computational efficiency.
Conclusion: Since uneven sampling occurs often
in biological measurements, the robust methods developed in this paper
are expected to have many uses. The regression based formulation of the
periodicity detection problem easily adapts to non-uniform sampling. Using
robust regression helps to reject inconsistently behaving data points.
Taesung
Park, Sung-Gon Yi, Seungmook Lee, Seung Yeoun Lee, Dong-Hyun Yoo, Jun-Ik
Ahn and Yong-Sung Lee
Motivation: Microarray technology allows the
monitoring of expression levels for thousands of genes simultaneously.
In time-course experiments in which gene expression is monitored over time,
we are interested in testing gene expression profiles for different experimental
groups. However, no sophisticated analytic methods have yet been proposed
to handle time-course experiment data.
Results: We propose a statistical test procedure
based on the ANOVA model to identify genes that have different gene expression
profiles among experimental groups in time-course experiments. Especially,
we propose a permutation test which does not require the normality assumption.
For this test, we use residuals from the ANOVA model only with time-effects.
Using this test, we detect genes that have different gene expression profiles
among experimental groups. The proposed model is illustrated using cDNA
microarrays of 3840 genes obtained in an experiment to search for changes
in gene expression profiles during neuronal differentiation of cortical
stem cells.
![]()
Kristof Engelen,
Bart Naudts, Bart De Moor and Kathleen Marchal
Motivation: We describe an approach to normalize
spotted microarray data, based on a physically motivated calibration model.
This model consists of two major components, describing the hybridization
of target transcripts to their corresponding probes on the one hand, and
the measurement of fluorescence from the hybridized, labeled target on
the other hand. The model parameters and error distributions are estimated
from external control spikes.
Results: Using a publicly available dataset,we
show that our procedure is capable of adequately removing the typical non-linearities
of the data, without making any assumptions on the distribution of differences
in gene expression from one biological sample to the next. Since our model
links target concentration to measured intensity, we show how absolute
expression values of target transcripts in the hybridization solution can
be estimated up to a certain degree.
Mugdha
Gadgil, Wei Lian, Chetan Gadgil, Vivek Kapur and Wei-Shou Hu
Background: DNA microarray is an invaluable
tool for gene expression explorations. In the two dye microarray, fluorescence
intensities of two samples, each labeled with a different dye, are compared
after hybridization. To compare a large number of samples, the 'reference
design' is widely used, in which all RNA samples are hybridized to a common
reference. Genomic DNA is an attractive candidate for use as a universal
reference, especially for bacterial systems with a low percentage of non-coding
sequences. However, genomic DNA, comprising of both the sense and anti-sense
strands, is unlike the single stranded cDNA usually used in microarray
hybridizations. The presence of the antisense strand in the 'reference'
leads to reactions between complementary labeled strands in solution and
may cause the assay result to deviate from true values.
Results: We have developed a mathematical
model to predict the validity of using genomic DNA as a reference in the
microarray assay. The model predicts that the assay can accurately estimate
relative concentrations for a wide range of initial cDNA concentrations.
Experimental results of DNA microarray assay using genomic DNA as a reference
correlated well to those obtained by a direct hybridization between two
cDNA samples. The model predicts that the initial concentrations of labeled
genomic DNA strands and immobilized strands, and the hybridization time
do not significantly affect the assay performance. At low values of the
rate constant for hybridization between immobilized and mobile strands,
the assay performance varies with the hybridization time and initial cDNA
concentrations. For the case where a microarray with immobilized single
strands is used, results from hybridizations using genomic DNA as a reference
will correspond to true ratios under all conditions.
Conclusion: Simulation using the mathematical
model, and the experimental study presented here show the potential utility
of microarray assays using genomic DNA as a reference. We conclude that
the use of genomic DNA as reference DNA should greatly facilitate comparative
transcriptome analysis.
V. Vinciotti,
R. Khanin, D. DAlimonte, X. Liu, N. Cattini, G. Hotchkiss, G. Bucca, O.
de Jesus, J. Rasaiyaah, C. P. Smith, P. Kellam and E. Wit
Motivation: Despite theoretical arguments that
so-called loop designs for two-channel DNA microarray experiments are
more efficient, biologists continue to use reference designs. We describe
two sets of microarray experiments withRNAfrom two different biological
systems (TPA-stimulated mammalian cells and Streptomyces coelicolor). In
each case, both a loop and a reference design were used with the same RNA
preparations with the aim of studying their relative efficiency.
Results: The results of these experiments
show that (1) the loop design attains a much higher precision than the
reference design, (2) multiplicative spot effects are a large source of
variability, and if they are not accounted for in the mathematical model,
for example, by taking log-ratios or including spot effects, then the model
will perform poorly. The first result is reinforced by a simulation study.
Practical recommendations are given on how simple loop designs can be extended
to more realistic experimental designs and how standard statistical methods
allow the experimentalist to use and interpret the results from loop designs
in practice.
![]()
M. Kathleen
Kerr, Mitchell Martin and Gary A. Churchill
Spotted
cDNA microarrays are emerging as a powerful and cost-effective tool for
largescale analysis of gene expression. Microarrays can be used to measure
the relative quantities of specific mRNAs in two or more tissue samples
for thousands of genes simultaneously. While the power of this technology
has been recognized, many open questions remain about appropriate analysis
of microarray data. One question is how to make valid estimates of the
relative expression for genes that are not biased by ancillary sources
of variation. Recognizing that there is inherent noise in microarray
data, how does one estimate the error variation associated with an estimated
change in expression, i.e., how does one construct the error bars? We demonstrate
that ANOVA methods can be used to normalize microarray data and provide
estimates of changes in gene expression that are corrected for potential
confounding effects. This approach establishes a framework for the general
analysis and interpretation of microarray data.
Wotao
Yin, Terrence Chen, Xiang Sean Zhou and Amit Chakraborty
Motivation: Background correction is an important
preprocess in cDNA microarray data analysis. A variety of methods have
been used for this purpose. However, many kinds of backgrounds, especially
inhomogeneous ones, cannot be estimated correctly using any of the existing
methods. In this paper, we propose the use of the TV+L1 model,
which minimizes the total variation (TV) of the image subject to an L1-fidelity
term, to correct background bias. We demonstrate its advantages over the
existing methods by both analytically discussing its properties and numerically
comparing it with morphological opening.
Results: Experimental results on both synthetic
data and real microarray images demonstrate that the TV+L1 model
gives the restored intensity that is closer to the true data than morphological
opening. As a result, this method can serve an important role in the preprocessing
of cDNA microarray data.
Henrik
Bengtsson, Göran Jönsson and Johan Vallon-Christersson
Background: Non-linearities in observed log-ratios
of gene expressions, also known as intensity dependent log-ratios, can
often be accounted for by global biases in the two channels being compared.
Any step in a microarray process may introduce such offsets and in this
article we study the biases introduced by the microarray scanner and the
image analysis software.
Results: By scanning the same spotted oligonucleotide
microarray at different photomultiplier tube (PMT) gains, we have identified
a channel-specific bias present in two-channel microarray data. For the
scanners analyzed it was in the range of 1525 (out of 65,535). The observed
bias was very stable between subsequent scans of the same array although
the PMT gain was greatly adjusted. This indicates that the bias does not
originate from a step preceding the scanner detector parts. The bias varies
slightly between arrays. When comparing estimates based on data from the
same array, but from different scanners, we have found that different scanners
introduce different amounts of bias. So do various image analysis methods.
We propose a scanning protocol and a constrained affine model that allows
us to identify and estimate the bias in each channel. Backward transformation
removes the bias and brings the channels to the same scale. The result
is that systematic effects such as intensity dependent log-ratios are removed,
but also that signal densities become much more similar. The average scan,
which has a larger dynamical range and greater signal-to-noise ratio than
individual scans, can then be obtained.
Conclusions: The study shows that microarray
scanners may introduce a significant bias in each channel. Such biases
have to be calibrated for, otherwise systematic effects such as intensity
dependent log-ratios will be observed. The proposed scanning protocol and
calibration method is simple to use and is useful for evaluating scanner
biases or for obtaining calibrated measurements with extended dynamical
range and better precision. The cross-platform R package aroma, which implements
all described methods, is available for free from http://www.maths.lth.se/
bioinformatics/.
![]()
K. K.
Dobbin, E. S. Kawasaki, D. W. Petersen and R. M. Simon
Motivation: Spot intensity serves as a proxy
for gene expression in dual-label microarray experiments. Dye bias is defined
as an intensity difference between samples labeled with different dyes
attributable to the dyes instead of the gene expression in the samples.
Dye bias that is not removed by array normalization can introduce bias
into comparisons between samples of interest. But if the bias is consistent
across samples for the same gene, it can be corrected by proper experimental
design and analysis. If the dye bias is not consistent across samples for
the same gene, but is different for different samples, then removing the
bias becomes more problematic, perhaps indicating a technical limitation
to the ability of fluorescent signals to accurately represent gene expression.
Thus, it is important to characterize dye bias to determine: (1) whether
it will be removed for all genes by array normalization, (2) whether it
will not be removed by normalization but can be removed by proper experimental
design and analysis and (3) whether dye bias correction is more problematic
than either of these and is not easily removable.
Results: We analyzed two large (each >27
arrays) tissue culture experiments with extensive dye swap arrays to better
characterize dye bias. Indirect, amino-allyl labeling was used in both
experiments. We found that post-normalization dye bias that is consistent
across samples does appear to exist for many genes, and that controlling
and correcting for this type of dye bias in design and analysis is advisable.
The extent of this type of dye bias remained unchanged under a wide range
of normalization methods (median-centering, various loess normalizations)
and statistical analysis techniques (parametric, rank based, permutation
based, etc.). We also found dye bias related to the individual samples
for a much smaller subset of genes. But these sample-specific dye biases
appeared to have minimal impact on estimated gene-expression differences
between the cell lines.
Catalin
C Barbacioru, Yulei Wang, Roger D Canales, Yongming A Sun, David N Keys,
Frances Chan, Karen A Poulter and Raymond R Samaha
Background: DNA microarray technology provides
a powerful tool for characterizing gene expression on a genome scale. While
the technology has been widely used in discovery-based medical and basic
biological research, its direct application in clinical practice and regulatory
decision-making has been questioned. A few key issues, including the reproducibility,
reliability, compatibility and standardization of microarray analysis and
results, must be critically addressed before any routine usage of microarrays
in clinical laboratory and regulated areas can occur. In this study we
investigate some of these issues for the Applied Biosystems Human Genome
Survey Microarrays.
Results: We analyzed the gene expression
profiles of two samples: brain and universal human reference (UHR), a mixture
of RNAs from 10 cancer cell lines, using the Applied Biosystems Human Genome
Survey Microarrays. Five technical replicates in three different sites
were performed on the same total RNA samples according to manufacturer's
standard protocols. Five different methods, quantile, median, scale, VSN
and cyclic loess were used to normalize AB microarray data within each
site. 1,000 genes spanning a wide dynamic range in gene expression levels
were selected for real-time PCR validation. Using the TaqMan® assays data
set as the reference set, the performance of the five normalization methods
was evaluated focusing on the following criteria: (1) Sensitivity and reproducibility
in detection of expression; (2) Fold change correlation with real-time
PCR data; (3) Sensitivity and specificity in detection of differential
expression; (4) Reproducibility of differentially expressed gene lists.
Conclusion: Our results showed a high level
of concordance between these normalization methods. This is true, regardless
of whether signal, detection, variation, fold change measurements and reproducibility
were interrogated. Furthermore, we used TaqMan® assays as a reference,
to generate TPR and FDR plots for the various normalization methods across
the assay range. Little impact is observed on the TP and FP rates in detection
of differentially expressed genes. Additionally, little effect was observed
by the various normalization methods on the statistical approaches analyzed
which indicates a certain robustness of the analysis methods currently
in use in the field, particularly when used in conjunction with the Applied
Biosystems Gene Expression System.
Marie-Laure Martin-Magniette, Julie Aubert, Eric Cabannes and Jean-Jacques Daudin
Motivation: In cDNA microarray experiments all
samples are labeled with either Cy3 or Cy5. Systematic and gene-specific
dye bias effects have been observed in dual-color experiments. In contrast
to systematic effects which can be corrected by a normalization method,
the gene-specific dye bias is not completely suppressed and may alter the
conclusions about the differentially expressed genes.
Methods: The gene-specific dye bias is taken
into account using an analysis of variance model. We propose an index,
named label bias index, to measure the gene-specific dye bias. It requires
at least two selfself hybridization cDNA microarrays.
Results: After lowess normalization we have
found that the gene specific dye bias is the major source of experimental
variability between replicates. The ratio (R/G) may exceed 2. As a consequence
false positive genes may be found in direct comparison without dye-swap.
The stability of this artifact and its consequences on gene variance and
on direct or indirect comparisons are addressed.
![]()
Kevin
K. Dobbin, Joanna H. Shih and Richard M. Simon
We show
here that the recommendations of Martin-Magniette et al. are fundamentally
flawed, and that in most realistic situations performing extensive dye-swap
arrays results in a poor experimental design. The key error made by these
authors is that they focus on oversimplified situations in which only two
RNA samples are being compared.
M.-L.
Martin-Magniette, J. Aubert, E. Cabannes and J.-J. Daudin
Thomas
Tang, Nicolas François, Annie Glatigny, Nicolas Agier, Marie-Hélène Mucchielli,
Lawrence Aggerbeck and Hervé Delacroix
Motivation: Two-colour microarrays are widely
used to perform transcriptome analysis. In most cases, it appears that
the red and green images resulting from the scan of a microarray slide
are slightly shifted one with respect to the other. To increase the robustness
of the measurement of the fluorescent emission intensities, multiple acquisitions
with the same or different PMT gains can be used. In these cases, a systematic
correction of image shift is required.
Results: To accurately detect this shift,
we first developed an approach using cross-correlation. Second, we evaluated
the most appropriate interpolation method to be used to derive the registered
image. Then, we quantified the effects of image shifts on spot quality,
using two different quality estimators. Finally, we measured the benefits
associated with a systematic image registration. In this study, we demonstrate
that registering the two images prior to data extraction provides a more
reliable estimate of the two colours ratio and thus increases the accuracy
of measurements of variations in gene expression.
Kathleen
F Kerr
Background: As part of its broad and ambitious
mission, the MicroArray Quality Control (MAQC) project reported the results
of experiments using External RNA Controls (ERCs) on five microarray platforms.
For most platforms, several different methods of data processing were considered.
However, there was no similar consideration of different methods for processing
the data from the Agilent two-color platform. While this omission is understandable
given the scale of the project, it can create the false impression that
there is consensus about the best way to process Agilent two-color data.
It is also important to consider whether ERCs are representative of all
the probes on a microarray.
Results: A comparison of different methods
of processing Agilent two-color data shows substantial differences among
methods for low-intensity genes. The sensitivity and specificity for detecting
differentially expressed genes varies substantially for different methods.
Analysis also reveals that the ERCs in the MAQC data only span the upper
half of the intensity range, and therefore cannot be representative of
all genes on the microarray.
Conclusion: Although ERCs demonstrate good agreement
between observed and expected logratios on the Agilent two-color platform,
such an analysis is incomplete. Simple loess normalization outperformed
data processing with Agilent's Feature Extraction software for accurate
identification of differentially expressed genes. Results from studies
using ERCs should not be overgeneralized when ERCs are not representative
of all probes on a microarray.
![]()
Henrik
Bengtsson and Ola Hössjer
Background: Low-level processing and normalization
of microarray data are most important steps in microarray analysis, which
have profound impact on downstream analysis. Multiple methods have been
suggested to date, but it is not clear which is the best. It is therefore
important to further study the different normalization methods in detail
and the nature of microarray data in general.
Results: A methodological study of affine
models for gene expression data is carried out. Focus is on two-channel
comparative studies, but the findings generalize also to single- and multi-channel
data. The discussion applies to spotted as well as in-situ synthesized
microarray data. Existing normalization methods such as curve-fit ("lowess")
normalization, parallel and perpendicular translation normalization, and
quantile normalization, but also dye-swap normalization are revisited in
the light of the affine model and their strengths and weaknesses are investigated
in this context. As a direct result from this study, we propose a robust
non-parametric multi-dimensional affine normalization method, which can
be applied to any number of microarrays with any number of channels either
individually or all at once. A high-quality cDNA microarray data set with
spike-in controls is used to demonstrate the power of the affine model
and the proposed normalization method.
Conclusion: We find that an affine model can
explain non-linear intensity-dependent systematic effects in observed log-ratios.
Affine normalization removes such artifacts for non-differentially expressed
genes and assures that symmetry between negative and positive log-ratios
is obtained, which is fundamental when identifying differentially expressed
genes. In addition, affine normalization makes the empirical distributions
in different channels more equal, which is the purpose of quantile normalization,
and may also explain why dye-swap normalization works or fails. All methods
are made available in the aroma package, which is a platform-independent
package for R.
Andy
G Lynch, David E Neal, John D Kelly, Glyn J Burtt and Natalie P Thorne
Background: There are mechanisms, notably ozone
degradation, that can damage a single channel of two-channel microarray
experiments. Resulting analyses therefore often choose between the unacceptable
inclusion of poor quality data or the unpalatable exclusion of some (possibly
a lot of) good quality data along with the bad. Two such approaches would
be a single channel analysis using some of the data from all of the arrays,
and an analysis of all of the data, but only from unaffected arrays. In
this paper we examine a 'combined' approach to the analysis of such affected
experiments that uses all of the unaffected data.
Results: A simulation experiment shows that
while a single channel analysis performs relatively well when the majority
of arrays are affected, and excluding affected arrays performs relatively
well when few arrays are affected (as would be expected in both cases),
the combined approach outperforms both. There are benefits to actively
estimating the key-parameter of the approach, but whether these compensate
for the increased computational cost and complexity over just setting that
parameter to take a fixed value is not clear. Inclusion of ozone-affected
data results in poor performance, with a clear spatial effect in the damage
being apparent.
Conclusion: There is no need to exclude unaffected
data in order to remove those which are damaged. The combined approach
discussed here is shown to out-perform more usual approaches, although
it seems that if the damage is limited to very few arrays, or extends to
very nearly all, then the benefits will be limited. In other circumstances
though, large improvements in performance can be achieved by adopting such
an approach.
Neil
D. Lawrence, Marta Milo, Mahesan Niranjan, Penny Rashbass and Stephan Soullier
Motivation: Gene expression levels are obtained
from microarray experiments through the extraction of pixel intensities
from a scanned image of the slide. It is widely acknowledged that variabilities
can occur in expression levels extracted from the same images by different
users with the same software packages. These inconsistencies arise due
to differences in the refinement of the placement of the microarray grids.
We introduce a novel automated approach to the refinement of grid placements
that is based upon the use of Bayesian inference for determining the size,
shape and positioning of the microarray spots, capturing uncertainty
that can be passed to downstream analysis.
Results: Our experiments demonstrate that
variability between users can be significantly reduced using the approach.
The automated nature of the approach also saves hours of researcherstime
normally spent in refining the grid placement.
![]()
Marianna
Zahurak, Giovanni Parmigiani, Wayne Yu, Robert B Scharpf, David Berman,
Edward Schaeffer, Shabana Shabbeer and Leslie Cope
Background: Pre-processing methods for two-sample
long oligonucleotide arrays, specifically the Agilent technology, have
not been extensively studied. The goal of this study is to quantify some
of the sources of error that affect measurement of expression using Agilent
arrays and to compare Agilent's Feature Extraction software with pre-processing
methods that have become the standard for normalization of cDNA arrays.
These include log transformation followed by loess normalization with or
without background subtraction and often a between array scale normalization
procedure. The larger goal is to define best study design and pre-processing
practices for Agilent arrays, and we offer some suggestions.
Results: Simple loess normalization without
background subtraction produced the lowest variability. However, without
background subtraction, fold changes were biased towards zero, particularly
at low intensities. ROC analysis of a spike-in experiment showed that differentially
expressed genes are most reliably detected when background is not subtracted.
Loess normalization and no background subtraction yielded an AUC of 99.7%
compared with 88.8% for Agilent processed fold changes. All methods performed
well when error was taken into account by t- or z-statistics, AUCs = 99.8%.
A substantial proportion of genes showed dye effects, 43% (99%CI : 39%,
47%). However, these effects were generally small regardless of the pre-processing
method.
Conclusion: Simple loess normalization without
background subtraction resulted in low variance fold changes that more
reliably ranked gene expression than the other methods. While t-statistics
and other measures that take variation into account, including Agilent's
z-statistic, can also be used to reliably select differentially expressed
genes, fold changes are a standard measure of differential expression for
exploratory work, cross platform comparison, and biological interpretation
and can not be entirely replaced. Although dye effects are small for most
genes, many array features are affected. Therefore, an experimental design
that incorporates dye swaps or a common reference could be valuable.
Figure 1. Exemple de plan d'expérience complet équilibré
* Les répétitions techniques correspondent à deux options d'un protocole de marquage (fluorochromes Cy3 vs Cy5) ou aux deux dépôts d'un gène sur la puce à ADN.
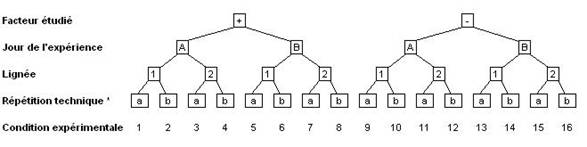
Figure 2. Effets du réglage du scanner sur la qualité des mesures
Dans cette expérience, les conditions + et - sont marquées avec deux fluorochromes différents. La Figure 2 gauche a été obtenue avec un réglage optimal du photomultiplicateur. La Figure 2 droite correspond à la même puce à ADN scannée avec un mauvais réglage du photomultiplicateur. La déformation du nuage de points est caractéristique d'une sensibilité insuffisante du photomultiplicateur pour un des deux fluorochromes. Dans ces figures, chaque point correspond à un gène. Un gène a pour abscisse son niveau d'expression moyen dans l'expérience et pour ordonnée le changement de niveau d'expression entre les conditions + et - (c'est-à-dire la différence des expressions moyennes dans les condition + et -). Le niveau d'expression est mesuré par le log de l'intensité du signal lu par le photomultiplicateur.
D'après Heidi Lyng et al. BMC Genomics 2004, 5 :10 ( http://www.biomedcentral.com/1471-2164/5/10 )
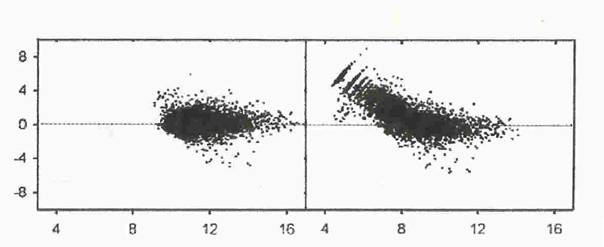
![]()
Figure 3 Effets du passage aux log après la correction du bruit de fond
Les figures représentent l'expression des gènes dans deux conditions expérimentales. Chaque condition a été normalisée, c'est-à-dire que l'intensité mesurée par le photomultiplicateur a subi une transformation logarithmique puis une transformation linéaire de telle sorte que la moyenne du log des intensités soit égale à 0 et la variance à 1.
Gauche : données initiales x j (min = 160)
Milieu : données corrigées x j - 160 + 0,01
Droite : introduction d'un seuil x j - 230 + 1 si x j > 230 autrement 1
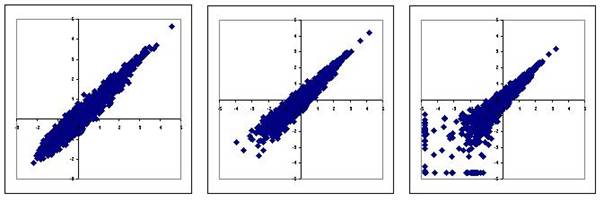
Figure 4 Comparaison des p-values obtenues par différents tests statistiques réalisés sur les mêmes données. La réalité des changements de niveau d'expression a été confirmée expérimentalement pour les gènes noirs.
Le signal est le même dans les trois tests, c'est l'écart entre l'expression dans la condition + et la condition -. Par contre les tests diffèrent dans la façon d'estimer le bruit :
• Analyse de variance (ANOVA). Le bruit correspond aux interactions entre les facteurs ;
• paired t-test (test t de Student pour des mesures appariées). Le bruit correspond à la variance lorsqu'on compare les conditions + et - appariées, c'est-à-dire toutes choses égales par ailleurs (les états sont les mêmes pour tous les autres facteurs) ;
• t-test (test t de Student). Le bruit correspond à la variance de l'ensemble des mesures faites sur le gène étudié.
Les méthodes les plus sensibles sont le paired t-test et l'ANOVA.
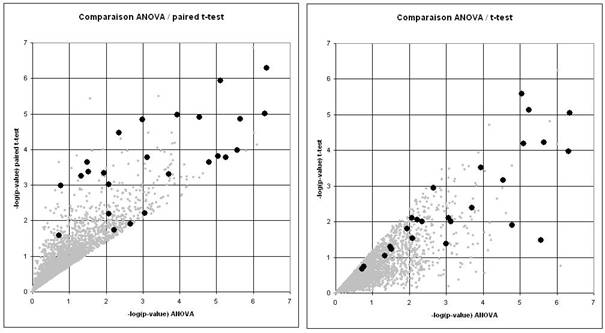
![]()
Figure 5 Représentation graphique des résultats d'une expérience.
Un gène a pour abscisse son niveau d'expression moyen dans l'expérience et pour ordonnée le changement de niveau d'expression entre les conditions + et - (c'est-à-dire la différence des expressions moyennes dans les condition + et -). La réalité des changements de niveau d'expression a été confirmée expérimentalement pour les gènes noirs.
Dans la Figure 3 haut, la moyenne est calculée en donnant le même poids à toutes les conditions expérimentales. Dans les deux autres figures il s'agit de moyennes pondérées. Dans la Figure 3 milieu, les poids sont calculés pour maximiser le carré de l'écart à la moyenne - la variance (Analyse en composantes principales ou ACP). Dans la Figure 3 bas, les poids sont calculés pour maximiser la puissance 4 de l'écart à la moyenne - le kurtosis (Analyse en composantes indépendantes ou ACI).
Nota : les deux axes ne sont pas à la même échelle afin d'augmenter la lisibilité des ordonnées.
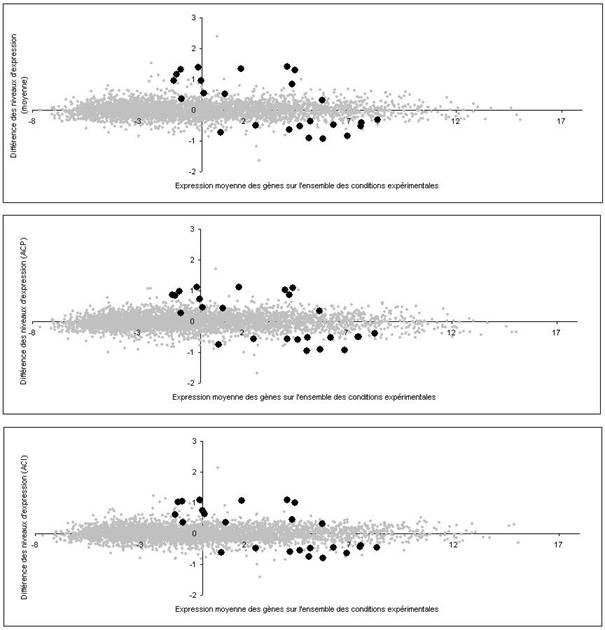
![]()
Figure 6 Comparaison de deux arbres représentant le même tableau de données (les distances entre les villes en France), mais construits avec des critères légèrement différents.
L'arbre de gauche minimise le carré des distances (critère de Ward) et l'arbre de droite la moyenne des distances (UPGMA). Bien que les deux arbres soient globalement satisfaisant, le groupe Nancy-Strasbourg n'est pas du tout à la même place dans les deux cas.
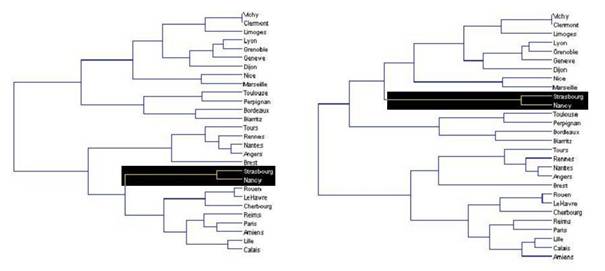
Figure 7 Analyse discriminante. Différents tracés possibles pour la frontière séparant les deux groupes de points. Dans l'image D, le tracé maximise la largeur de la marge qui sépare les deux groupes (méthode SVM : Support Vector Machines ou Séparatrices à Vaste Marge en français).
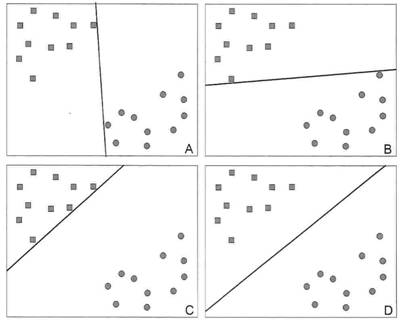
![]()
Encart 1. Estimation robuste de la moyenne et de l'écart-type
Ordonner les N observation par valeur croissante puis déterminer les valeurs :
xa = valeur de l'observation ayant le rang N × 1/16
xb = valeur de l'observation ayant le rang N × 1/4
xc = valeur de l'observation ayant le rang N × 1/2 (c'est-à-dire la médiane)
xd = valeur de l'observation ayant le rang N × 3/4
xe = valeur de l'observation ayant le rang N × 15/16
Calculer la moyenne
m = 1/6 ( xa + xb + 2 ×xc + xd + xe)
et l'écart-type
s = 1/4 ( xa + 3/4 ×xb - 3/4 ×xd - xe)
Selon M.H. Quenouille. Méthodes de calcul statistiques rapides . Dunod 1964.
Encart 2. Deux ou trois sites utiles
Le site de Bioconductor ( http://www.bioconductor.org/ ) contient un ensemble très complet d'outils statistiques adaptés à l'analyse du transcriptome.
Le site GEPAS ( http://gepas.bioinfo.cipf.es/ ) offre une suite logicielle en ligne qui permet au biologiste d'analyser ses données sans rien installer sur sa propre machine.
Un site d'autoformation à l'analyse statistique des expériences sur le transcriptome est disponible à l'Université de Tours ( cf. le sommaire de GENET à http://genet.univ-tours.fr//index.htm ).
Par ailleurs, l'analyse statistique peut être réalisée sans difficulté en couplant un tableur comme Excel et un des nombreux logiciels statistiques, spécialisés ou généralistes, existants actuellement ( cf . par exemple http://linus.nci.nih.gov/BRB-ArrayTools.html ou XLSTAT http://www.xlstat.com/fr/home/ ).
![]()

© Université
de TOURS - GÉNET
Document modifié le
25 mars, 2010
![]()
![]()
![]()
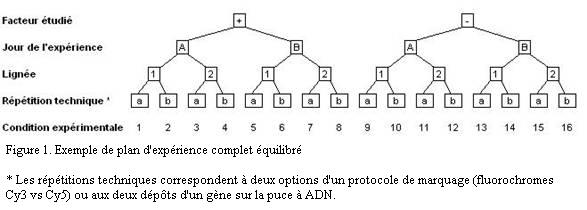{kind=link}
{kind=link}
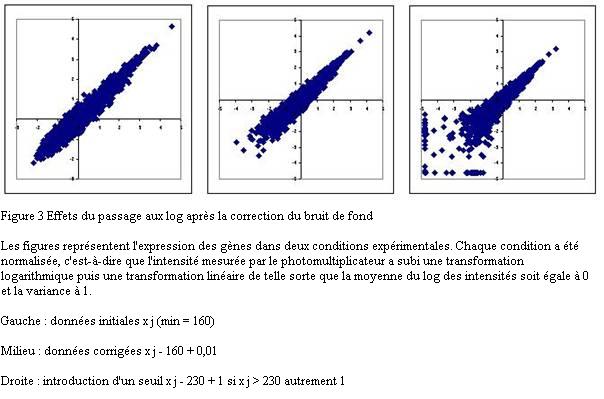{kind=link}
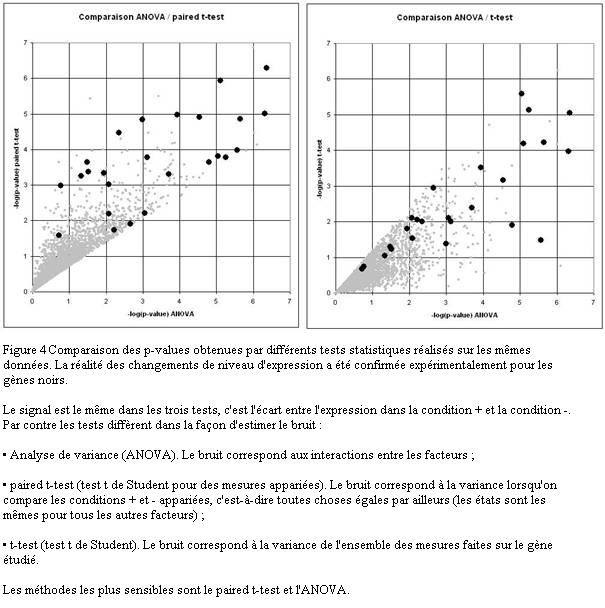{kind=link}
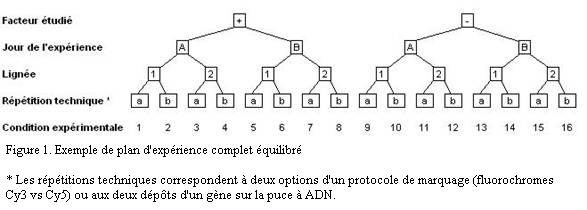{kind=link}
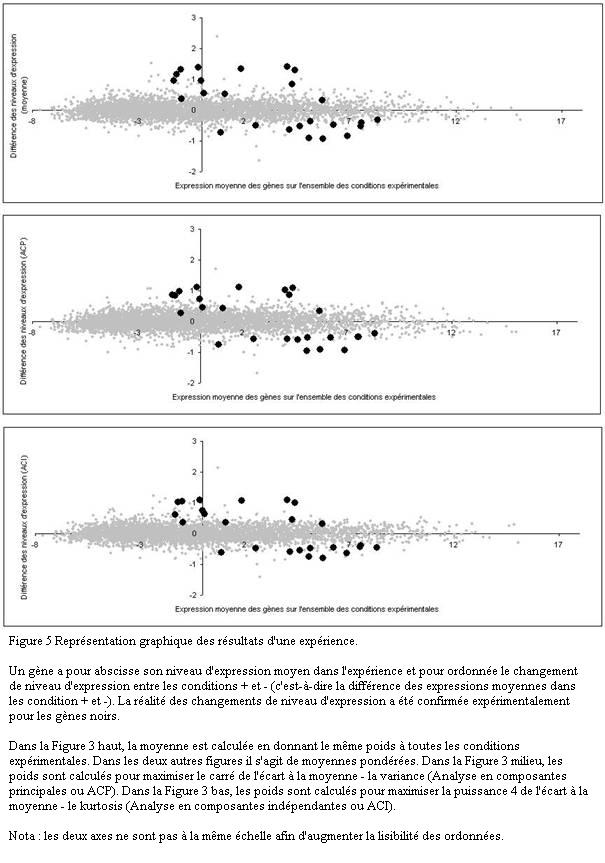{kind=link}
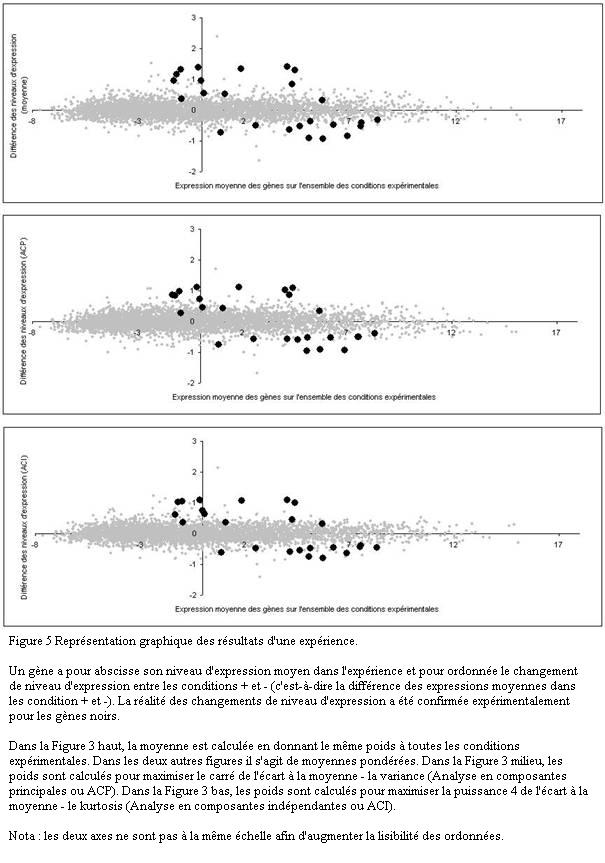{kind=link}
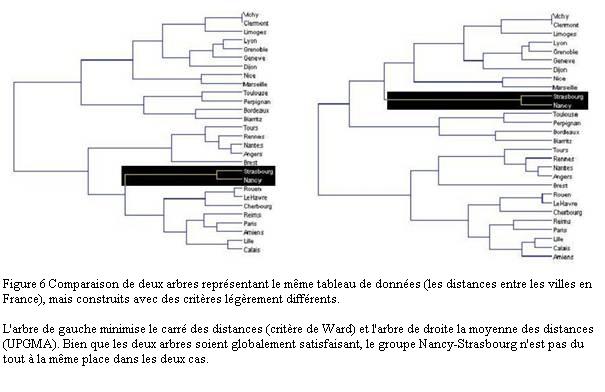{kind=link}
{kind=link}
{kind=link}